





Overcoming resistance to anti-PD-1/PD-L1 therapy in cancer
 0
0 Abstract
Recently, programmed death-1 (PD-1)/programmed death-ligand 1 (PD-L1) inhibitors have achieved major breakthroughs in oncology, with 32 drugs approved over the past decade. This advancement has established immunotherapy as the fifth major antitumor modality following surgery, chemotherapy, radiotherapy, and targeted therapy. However, PD-1/PD-L1 inhibitors induce sustained responses in only a limited number of patients, and primary and acquired resistance remain critical challenges in clinical practice. As understanding of the complex crosstalk among cancer cells, the tumor microenvironment, and the host immune system deepens, numerous strategies to overcome PD-1/PD-L1 inhibitor resistance have been proposed. In this review, we examine the current development of PD-1/PD-L1 inhibitors, analyze global approval trends, and evaluate their monotherapy efficacy across various tumor types. As multi-target combination therapy is an essential strategy for overcoming resistance, we analyze key combination targets - such as vascular endothelial growth factor (VEGF), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and lymphocyte activation gene 3 (LAG-3) - and highlight the clinical success of novel dual-target regimens such as ivonescimab (PD-1/VEGF). Furthermore, we discuss potential approaches to overcoming resistance from both microenvironmental (e.g., targeting cancer-associated fibroblasts or utilizing antibody-drug conjugates) and macroenvironmental (e.g., modulating the microbiota or sex hormones) perspectives. This review provides a forward-looking framework for designing precision- and mechanism-driven combination therapies aimed at converting non-responders into responders.
Keywords
INTRODUCTION
Immune checkpoint inhibitors (ICIs), particularly programmed death-1 (PD-1)/programmed death-ligand 1 (PD-L1) monoclonal antibodies, have markedly improved survival outcomes for cancer patients. Research on PD-1/PD-L1 began in 1992, when Ishida et al. cloned the complementary DNA (cDNA) of PD-1 from immune cell lines undergoing apoptosis[1]. Subsequently, a series of studies investigating the function and therapeutic potential of PD-1/PD-L1 emerged [Supplementary Figure 1]. Through backcrossing PD-1 knockout (KO) heterozygous mice with inbred lines and immunodeficient strains, Nishimura demonstrated how the absence of this receptor drives the activation of T lymphocytes[2]. In 1999, Dong et al. first identified B7 homolog 1 (B7-H1) by screening human cDNA libraries for homologous sequences and clarified its regulatory role in T cells[3]. Later, the interaction between PD-1 and B7-H1 (also known as PD-L1) was rapidly recognized[4]. The path from discovery to clinical translation has been extensively explored. An early obstacle was the discrepancy between laboratory and clinical samples: fresh tumor biopsies frequently displayed significant PD-L1 levels, whereas cancer cells grown in vitro typically lacked this ligand. In 2002, Dong et al. demonstrated that PD-L1 overexpression in tumors is primarily regulated by interferon gamma (IFN-γ)[5]. Most importantly, the study showed that anti-PD-L1 antibodies could restore T cell function and inhibit tumor growth in preclinical models. Subsequent studies by Iwai et al. and Curiel et al. further supported that blocking the interaction between PD-1 and its ligand could effectively suppress cancers[6,7]. Collectively, these findings underscored the critical role of the PD-1/PD-L1 pathway in tumor immune evasion and stimulated further investigation into its regulatory mechanisms, ranging from genomic alterations to protein-level modifications [Figure 1]. The transformative impact of this foundational research was recognized in 2018 with the Nobel Prize awarded to Prof. Honjo.
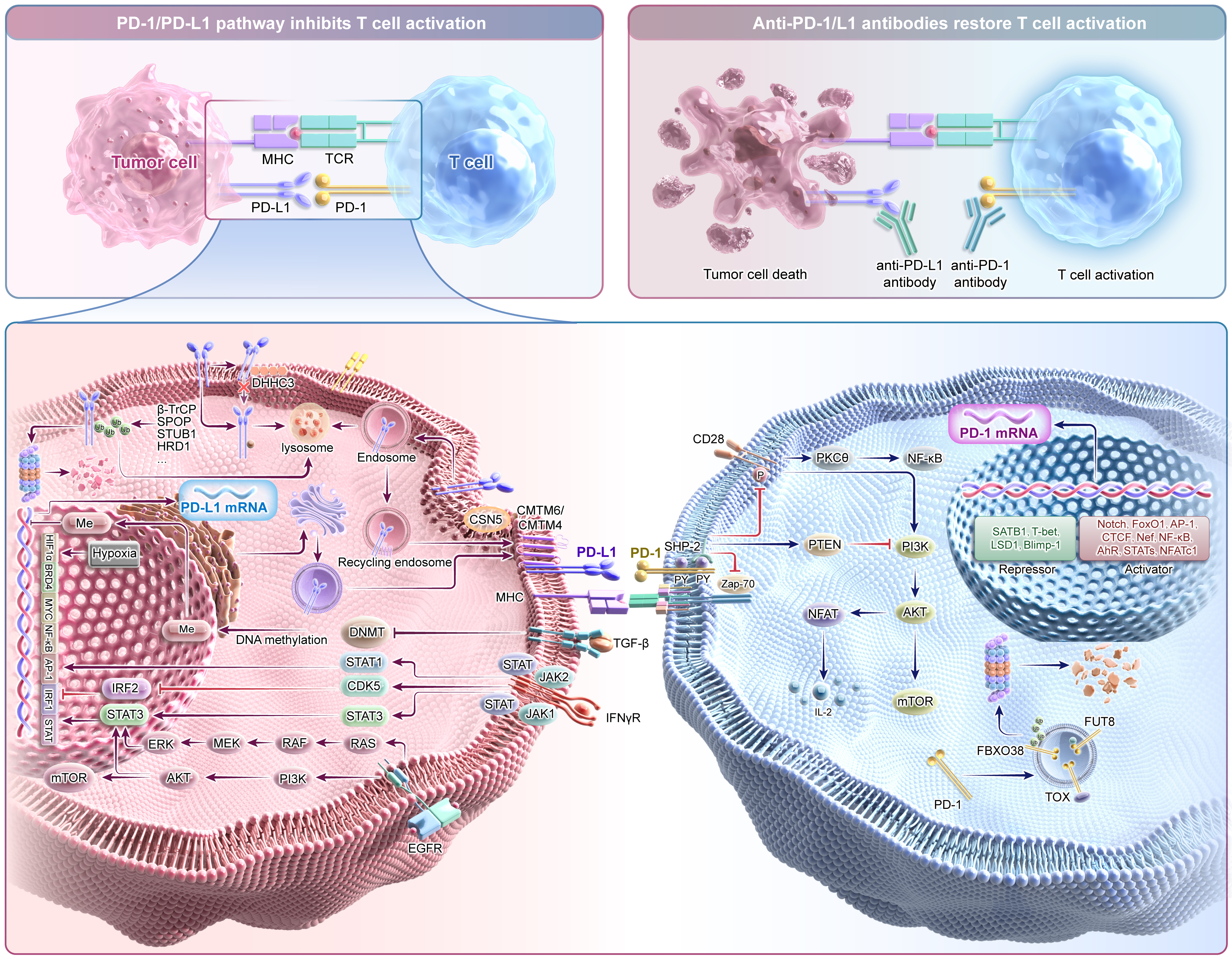
Figure 1. Regulatory network of the PD-1/PD-L1 signaling pathway. The interaction between PD-1 and PD-L1 inhibits TCR signaling, thereby suppressing T cell activation. Expression of this axis is modulated through diverse mechanisms, ranging from genomic alterations to targeted protein degradation. Blockade of the PD-1/PD-L1 axis with anti-PD-1/PD-L1 antibodies can relieve T cell suppression and promote tumor cell eradication. PD-1: Programmed death-1; PD-L1: programmed death-ligand 1; TCR: T cell receptor; MHC: major histocompatibility complex; DHHC3: palmitoyltransferase DHHC3; Ub: Ubiquitin; β-TrCP: beta-transducin repeat containing protein; SPOP: speckle-type POZ protein; STUB1: STIP1 homology and U-box containing protein 1; HRD1: HMG-CoA reductase degradation protein 1; CSN5: COP9 signalosome 5; CMTM: CKLF-like MARVEL transmembrane domain containing; TGF-β: transforming growth factor beta; DNMT: DNA methyltransferase; Me: methylation; IFNγR: interferon gamma receptor; JAK: Janus kinase; STAT: signal transducer and activator of transcription; CDK5: cyclin dependent kinase 5; IRF: interferon regulatory factor; EGFR: epidermal growth factor receptor; RAS: rat sarcoma virus oncogene homolog; RAF: RAF proto-oncogene serine/threonine-protein kinase; MEK: mitogen-activated protein kinase kinase; ERK: extracellular signal-regulated kinase; PI3K: phosphoinositide 3-kinase; AKT: AKT serine/threonine kinase; mTOR: mechanistic target of rapamycin; HIF-1α: hypoxia-inducible factor 1 subunit alpha; BRD4: bromodomain containing 4; MYC: MYC proto-oncogene; NF-κB: nuclear factor kappa B; AP-1: activator protein 1; SHP-2: SH2 domain-containing protein tyrosine phosphatase 2; Zap-70: zeta-chain-associated protein kinase 70; PKCθ: protein kinase C theta; PTEN: phosphatase and tensin homolog; NFAT: nuclear factor of activated T cells; IL-2: interleukin-2; SATB1: special AT-rich sequence-binding protein 1; T-bet: T-box transcription factor 21; LSD1: lysine specific demethylase 1; Blimp-1: B lymphocyte-induced maturation protein-1; FoxO1: forkhead box O1; CTCF: CCCTC-binding factor; AhR: aryl hydrocarbon receptor; NFATc1: nuclear factor of activated T cells 1; TOX: thymocyte selection-associated high mobility group box protein; FUT8: fucosyltransferase 8; FBXO38: F-box protein 38.
Since the first PD-1 inhibitor received U.S. Food and Drug Administration (FDA) approval in 2014, clinical applications of PD-1/PD-L1 blockade have expanded significantly across oncology. Compared with traditional treatment, PD-1/PD-L1 inhibitors have achieved higher disease control rates and improved overall survival (OS). However, poor responses are still observed in certain patient groups, and durable responses occur only in a subset of patients, even among tumors typically sensitive to ICIs. Therefore, primary or secondary resistance remains a major clinical challenge. As research continues to explore the multidimensional crosstalk among tumors, the immune system, and other biological systems, our comprehension of anti-PD-1/PD-L1 resistance mechanisms continues to evolve[8]. Tumor-intrinsic components, including tumor cells and their secretions, along with immune cells, stromal cells, and the microbiome, collectively serve as key determinants of therapeutic response to anti-PD-1/PD-L1 therapy. Tumor-intrinsic factors form the foundation of immune responses, while the tumor immune microenvironment shapes the overall immune landscape. Additionally, systemic host-related factors such as sex, obesity, and microbiota composition may also affect responses to anti-PD-1/PD-L1 therapy[9-11]. Beyond host-intrinsic influences, the external environment may further modulate cancer biology and potentially contribute to therapeutic resistance. Given that anti-tumor immunity is intricately regulated by both micro- and macro-environment factors, combination therapy may be an essential strategy to maximize the therapeutic potential of PD-1/PD-L1 blockade.
This article summarizes globally approved PD-1/PD-L1 inhibitors and outlines the current therapeutic landscape. We first analyzed objective response rates (ORRs) from 118 clinical trials, which included a total of 22,433 patients across different cancer types. To explore combination strategies for overcoming resistance, we retrieved and analyzed 7,381 PD-1/PD-L1-related clinical trials from ClinicalTrials.gov. Furthermore, this review synthesizes preclinical evidence underlying tumor immune escape during anti-PD-1/PD-L1 therapy, as well as actionable approaches to restoring therapeutic sensitivity.
APPROVAL OF ANTI-PD-1/PD-L1 DRUGS
A comprehensive search of ClinicalTrials.gov identified 7,381 clinical trials evaluating PD-1/PD-L1 blockade by April 2025 [Figure 2]. Globally, 32 PD-1/PD-L1 inhibitors have been approved for antitumor therapy, including 21 targeting PD-1 and 11 targeting PD-L1 [Figure 3]. Among these, 10 drugs were approved by the U.S. FDA, 19 by the Chinese National Medical Products Administration (NMPA), and the remaining three were approved in Japan, Europe, and Russia, respectively. Notably, the pace of development has accelerated markedly since 2021, with 21 drugs approved over the past 4 years. The first bispecific antibody targeting PD-1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), cadonilimab, received approval in 2022[12]. Two years later, the first bispecific antibody targeting PD-1 and vascular endothelial growth factor (VEGF), ivonescimab, was also approved[13]. Additionally, PD-1/lymphocyte activation gene 3 (LAG-3) or PD-1/CTLA-4 bifunctional combination antibodies (OPDUALAG, iparomlimab, and tuvonralimab) were approved in 2022 and 2024, respectively[14,15].
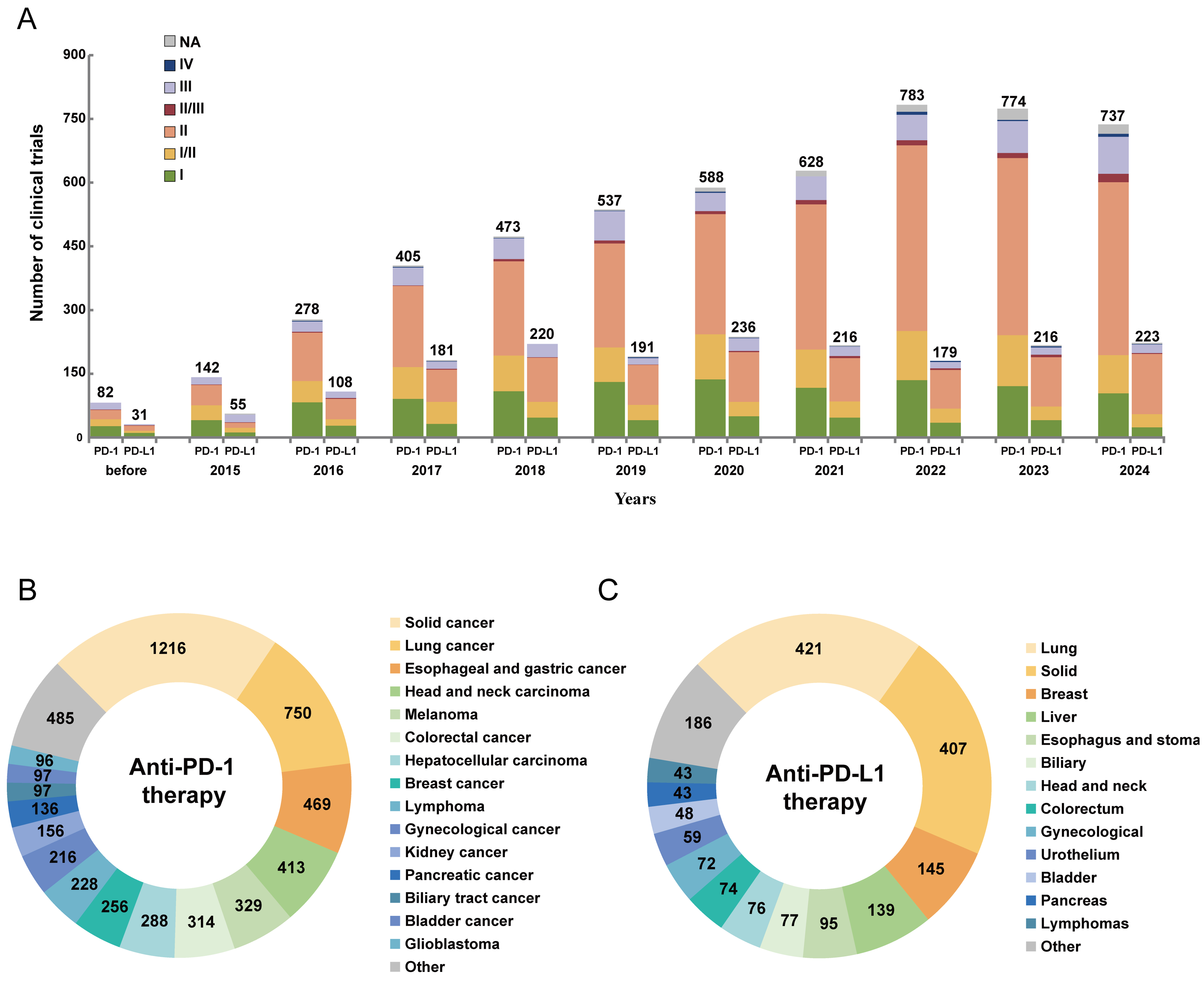
Figure 2. The development landscape of clinical trials related to PD-1/PD-L1 inhibitors. (A) Number of clinical trials involving PD-1/PD-L1 inhibitors across different phases; (B) Distribution of cancer types in clinical trials related to anti-PD-1 therapy; (C) Distribution of cancer types in clinical trials related to anti-PD-L1 therapy. Data from ClinicalTrials.gov. PD-1: Programmed death-1; PD-L1: programmed death-ligand 1; NA: not applicable.
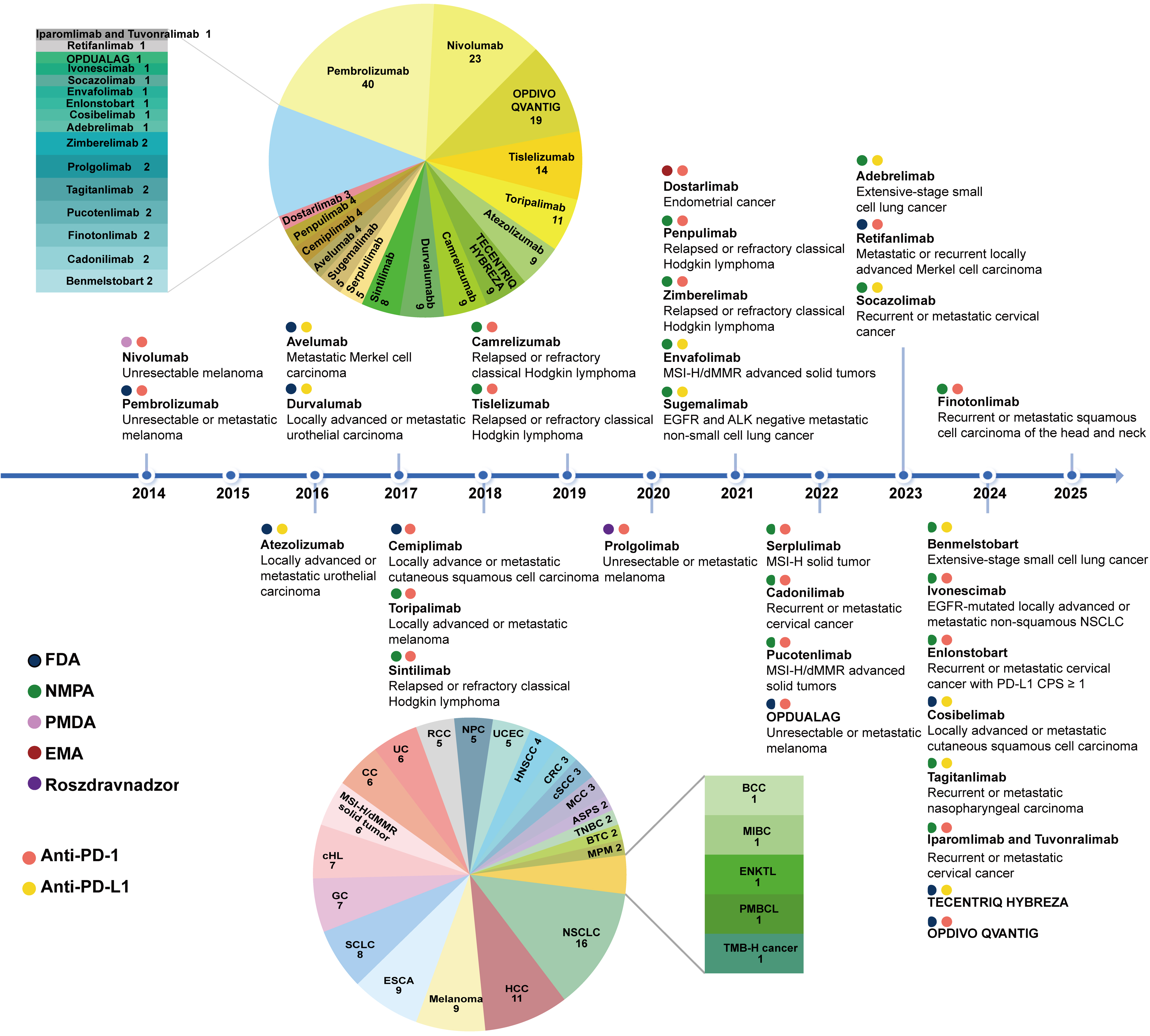
Figure 3. Timeline of PD-1/PD-L1 inhibitor approvals. The timeline illustrates the first regulatory agency to approve a PD-1/PD-L1 inhibitor and the indications for 32 PD-1/PD-L1 inhibitors. The pie chart in the upper left shows the number of approved indications for each drug. The pie chart in the lower left displays the number of available PD-1/PD-L1 inhibitors for each cancer type. PD-1: Programmed death-1; PD-L1: programmed death-ligand 1; MSI-H: microsatellite instability-high; dMMR: mismatch repair deficiency; EGFR: epidermal growth factor receptor; ALK: anaplastic lymphoma kinase; FDA: Food and Drug Administration; NMPA: National Medical Products Administration (China); PMDA: Pharmaceuticals and Medical Devices Agency (Japan); EMA: European Medicines Agency; Roszdravnadzor: Federal Service for Surveillance in Healthcare (Russia); NSCLC: non-small cell lung cancer; CPS: combined positive score; CC: cervical cancer; UC: urothelial cancer; RCC: renal cell carcinoma; NPC: nasopharyngeal carcinoma; UCEC: endometrial cancer; HNSCC: head and neck squamous cell cancer; CRC: colorectal cancer; cSCC: cutaneous squamous cell carcinoma; MCC: Merkel cell carcinoma; ASPS: alveolar soft part sarcoma; TNBC: triple-negative breast cancer; BTC: biliary tract cancer; MPM: malignant pleural mesothelioma; HCC: hepatocellular carcinoma; ESCA: esophageal cancer; SCLC: small cell lung cancer; GC: gastric cancer; cHL: classical Hodgkin lymphoma; BCC: basal cell carcinoma; MIBC: muscle-invasive bladder cancer; ENKTL: extranodal NK/T cell lymphoma; PMBCL: primary mediastinal large B-cell lymphoma; TMB-H: tumor mutational burden-high.
Malignant melanoma served as the initial clinical focus for PD-1/PD-L1 inhibitors. The therapeutic landscape subsequently expanded to encompass non-small cell lung cancer (NSCLC), nasopharyngeal carcinoma, and other malignancies [Figure 3]. Pembrolizumab has received approval for the widest range of indications, covering 40 indications across 20 cancer types, including malignant melanoma, NSCLC, and head and neck squamous cell carcinoma[16]. Atezolizumab is the first approved PD-L1 inhibitor and has nine indications across five cancer types[17]. NSCLC has the largest number of approved therapeutic options, including 11 PD-1 inhibitors, four PD-L1 inhibitors, and one PD-1/VEGF bispecific antibody[18,19]. This is followed by hepatocellular carcinoma (HCC), malignant melanoma, and esophageal cancer. Similarly, the exploration of indications in clinical trials broadly aligns with this trend [Figure 2B and C]. The drug development landscape corresponds well with tumor epidemiological characteristics, as both NSCLC and HCC rank among the top 10 cancers in incidence and mortality[20]. Currently, 32 approved PD-1/PD-L1 inhibitors are available for 26 cancer types, covering not only first- and later-line treatments for advanced disease but also adjuvant and neoadjuvant applications for early disease.
ANTI-PD-1/PD-L1 MONOTHERAPY EFFICACY AND COMBINATION STRATEGIES
Despite their broad application, single-agent PD-1/PD-L1 blockade has shown heterogeneous clinical outcomes across cancer types, and not all patients benefit[21]. To evaluate these variable response rates, we conducted a comprehensive literature search in PubMed, which initially retrieved 12,842 records [Supplementary Table 1 and Supplementary Figure 2]. After screening, 118 eligible clinical trials were included, involving 22,433 patients with advanced solid tumors [Figure 4 and Supplementary Table 2]. Overall, the data revealed that first-line administration of these monotherapies achieved ORRs ranging from 14.8% to 53%, whereas responses in later-line settings ranged from 2.3% to 48%. In tumors such as Merkel cell carcinoma, cutaneous squamous carcinoma, and microsatellite instability-high colorectal cancer, ORRs reached about 50% in the first-line setting and 30% in later lines[22,23]. However, in gastric and liver cancers, first-line ORRs were only around 15%[24]. Improving anti-PD-1/PD-L1 response therefore remains a critical challenge.
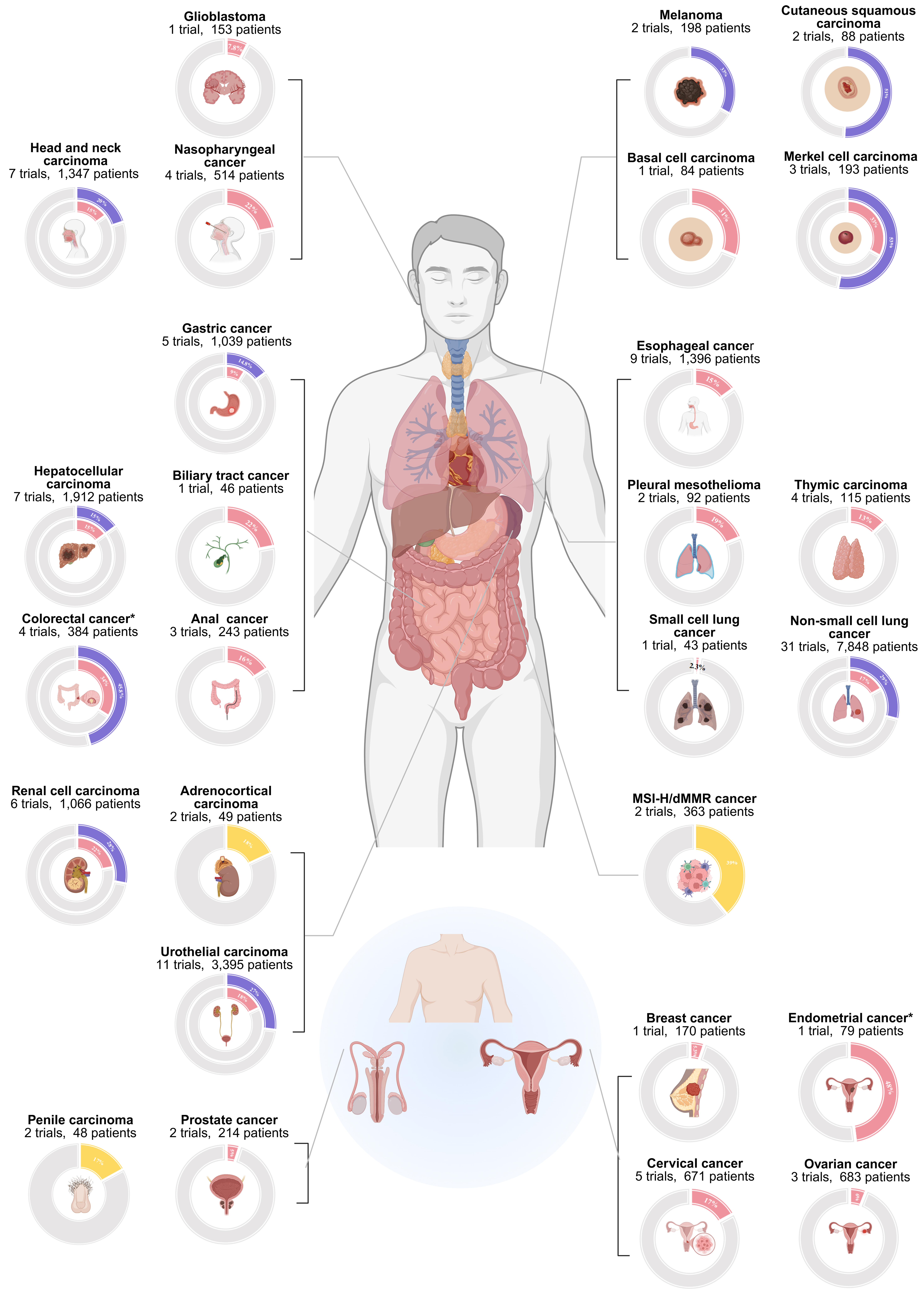
Figure 4. ORRs for PD-1/PD-L1 inhibitors as monotherapy. Purple circles show ORRs for PD-1/PD-L1 inhibitors as first-line treatment; pink circles show ORRs for their use in subsequent therapy; yellow circles show ORRs regardless of treatment line. *Tumors are microsatellite instability-high or mismatch repair-deficient. Created in BioRender. li, L. (2026) https://BioRender.com/kws7r1d. ORRs: Objective response rates; PD-1: programmed death-1; PD-L1: programmed death-ligand 1; MSI-H: microsatellite instability-high; dMMR: mismatch repair deficiency.
Combination therapies targeting multiple pathways to enhance antitumor activity are the main strategy to address this challenge. Analysis of 7,381 PD-1/PD-L1-related clinical trials showed that VEGF and CTLA-4 are the most common combination targets, with other major targets including poly (ADP-ribose) polymerase (PARP), human epidermal growth factor receptor 2 (HER2), LAG-3, and T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains (TIGIT) [Figure 5A]. Bispecific antibodies represent a promising direction[25]. Beyond the four approved PD-1-related bispecific or bifunctional agents, 12 additional drugs are currently in phase III clinical trials and may be approved in the near future [Figure 5B]. The antitumor mechanisms of these common combination targets are illustrated in Figure 5C. The addition of bevacizumab to anti-PD-1/PD-L1 therapy has been widely used in patients with cancers[26,27]. Recently, combinations with other anti-angiogenic agents, such as lenvatinib and axitinib, have also gained approval[28,29]. Notably, ivonescimab, a bispecific PD-1/VEGF antibody, demonstrated superior efficacy in a head-to-head comparison with pembrolizumab in the HARMONi-2 study. Interim data showed nearly doubled median progression-free survival (PFS) (11.1 vs. 5.8 months)[30]. Based on these findings, ivonescimab was approved in 2024 for the treatment of EGFR-mutated locally advanced or metastatic non-squamous NSCLC. Findings from the HARMONi-6 trial, which evaluated ivonescimab plus chemotherapy vs. tislelizumab plus chemotherapy in advanced squamous NSCLC, further confirmed its antitumor activity[31]. Combining PD-1/PD-L1 inhibitors with CTLA-4 inhibitors is one of the earliest dual immunotherapy strategies. Results from the phase III POSEIDON study showed that adding tremelimumab to durvalumab plus chemotherapy significantly improved OS and PFS in metastatic NSCLC without increasing adverse effects[32]. Findings from the CheckMate 9LA trial further supported the antitumor efficacy of this approach[33]. Co-targeting LAG-3 and the PD-1/PD-L1 axis represents another effective strategy. A phase II/III trial demonstrated that OPDUALAG, a fixed-dose combination of relatlimab and nivolumab, significantly prolonged survival in patients with advanced melanoma compared with nivolumab monotherapy[34]. However, not all combination regimens have shown positive outcomes. The first PD-L1/transforming growth factor beta (TGF-β) bifunctional fusion protein (M7824) entered clinical trials in 2015, but several phase II/III studies were terminated due to failure to meet primary endpoints, and no agents in this category have been approved to date[35,36]. Safety concerns have also emerged in certain combinations, such as nivolumab plus abemaciclib, as well as pembrolizumab or nivolumab plus pegilodecakin[37,38]. Overall, the exploration of combination strategies still faces substantial challenges, requiring continued preclinical research to clarify mechanisms of antitumor immunity and provide theoretical support for overcoming resistance.
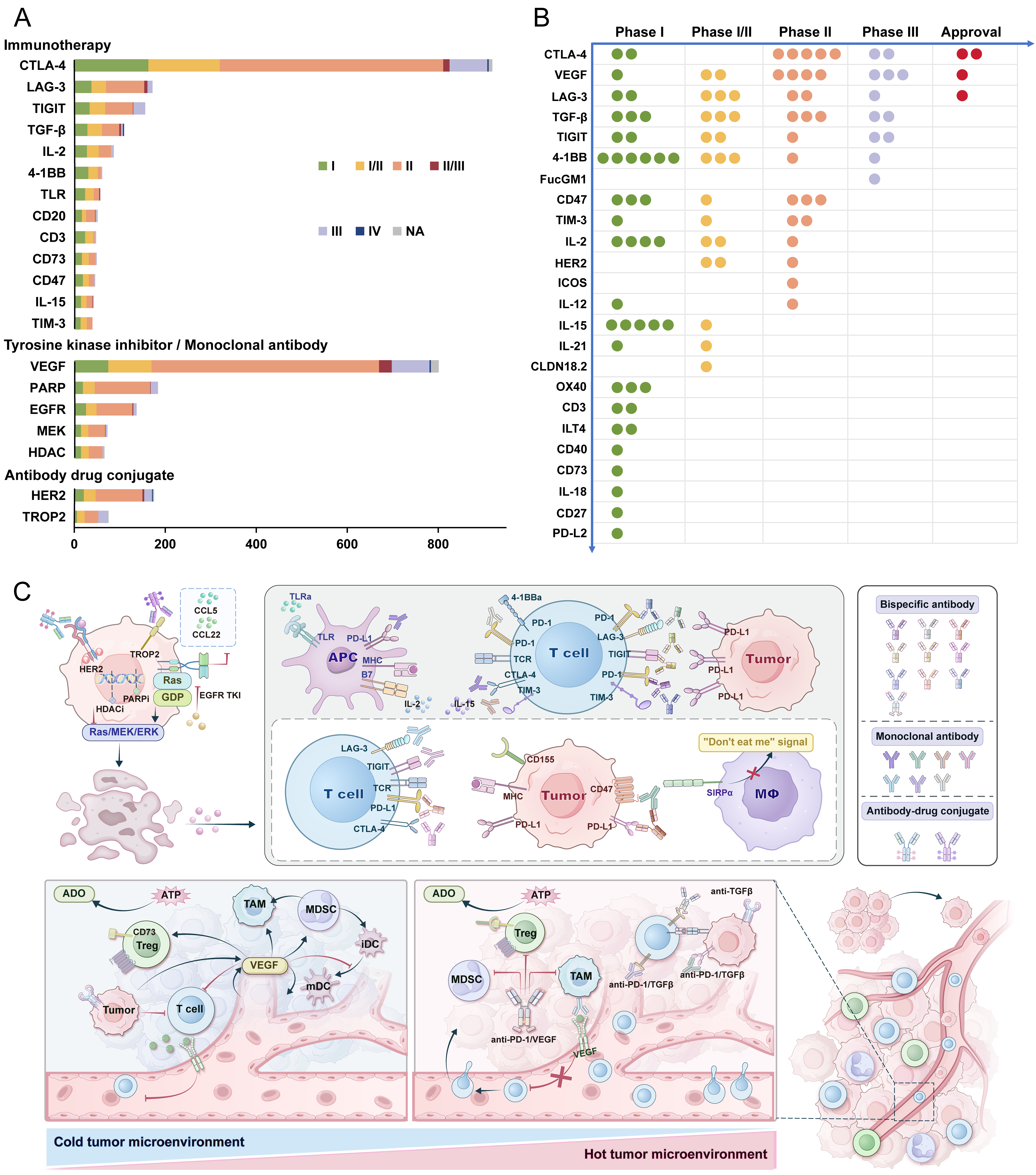
Figure 5. Drug targets combined with PD-1/PD-L1 inhibitors in clinical trials. (A) Top 20 combination therapy targets across 7,381 clinical trials; (B) PD-1/PD-L1-related bispecific or bifunctional combination antibodies in clinical trials or on the market, with each dot representing one drug; (C) Mechanisms of the most common combination therapy targets. PD-1: Programmed death-1; PD-L1: programmed death-ligand 1; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; LAG-3: lymphocyte activation gene 3; TIGIT: T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domains; TGF-β: transforming growth factor beta; IL: interleukin; TLR: Toll-like receptor; CD: cluster of differentiation; TIM-3: T cell immunoglobulin and mucin domain-containing protein 3; NA: not available; VEGF: vascular endothelial growth factor; PARP: poly (ADP-ribose) polymerase; EGFR: epidermal growth factor receptor; MEK: mitogen-activated protein kinase kinase; HDAC: histone deacetylase; HER2: human epidermal growth factor receptor 2; TROP2: trophoblast cell surface antigen 2; ICOS: inducible T-cell costimulator; CLDN: claudin; OX40: OX40 molecule; ILT4: immunoglobulin-like transcript 4; PD-L2: programmed death-ligand 2; CCL: C-C motif chemokine ligand; GDP: guanosine diphosphate; TKI: tyrosine kinase inhibitor; ERK: extracellular signal-regulated kinase; APC: antigen-presenting cell; MHC: major histocompatibility complex; TCR: T cell receptor; SIRPα: signal regulatory protein alpha; ADO: adenosine; ATP: adenosine triphosphate; Treg: regulatory T; TAM: tumor-associated macrophage; MDSC: myeloid-derived suppressor cell; iDC: immature dendritic cell; mDC: mature dendritic cell.
PRECLINICAL RESEARCH ON PD-1/PD-L1 INHIBITOR-BASED THERAPY
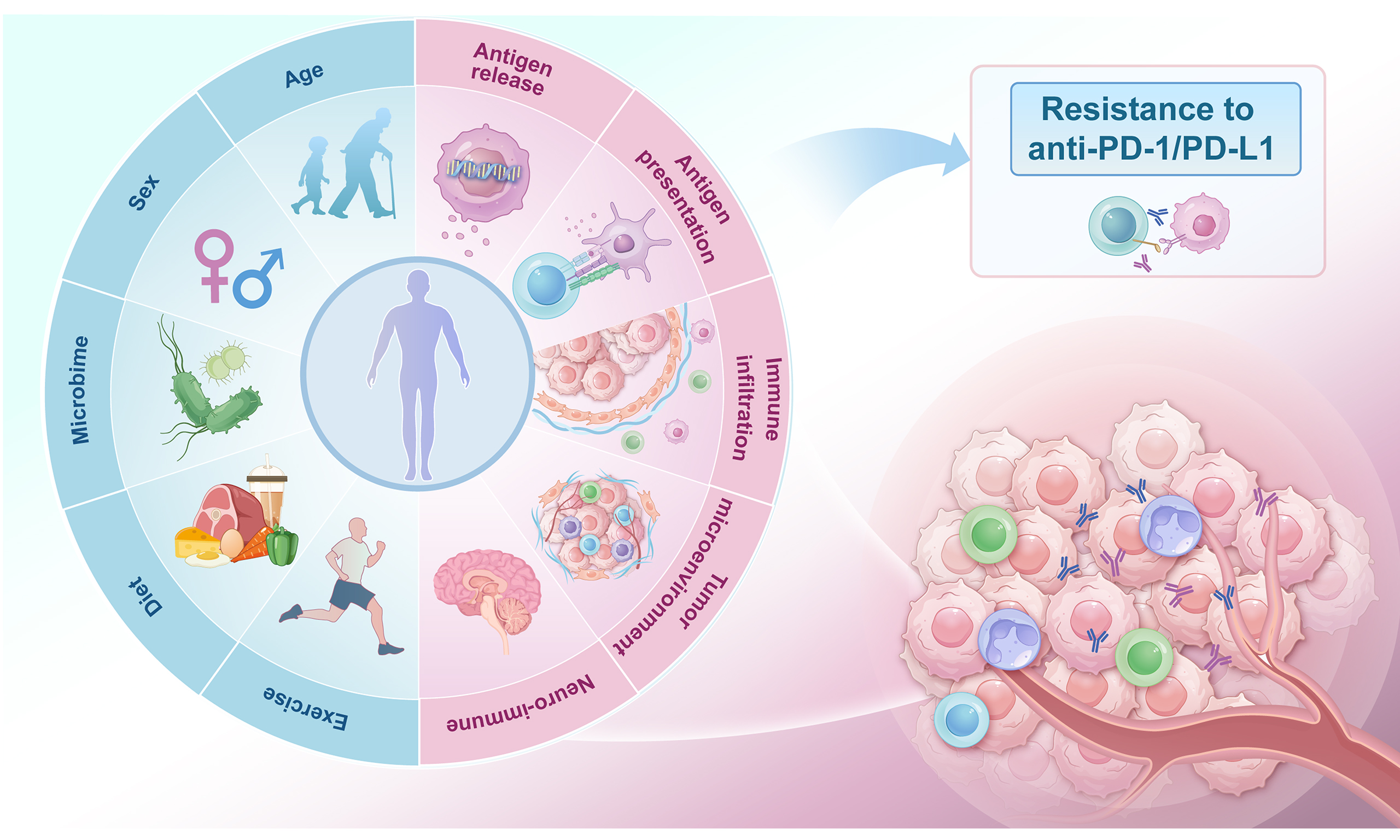
To clarify the preclinical landscape of anti-PD-1/PD-L1 resistance, we reviewed the relevant literature from PubMed and summarized potential strategies for restoring drug activity. Disruption at any stage of the cancer-immunity cycle can lead to immune escape, thereby impairing the potency of ICIs. Furthermore, the influence of host-related factors such as sex, age, and gut microbiota is increasingly recognized. This study examined resistance mechanisms from macro- and microenvironmental perspectives, providing theoretical insights into overcoming resistance [Figure 6].
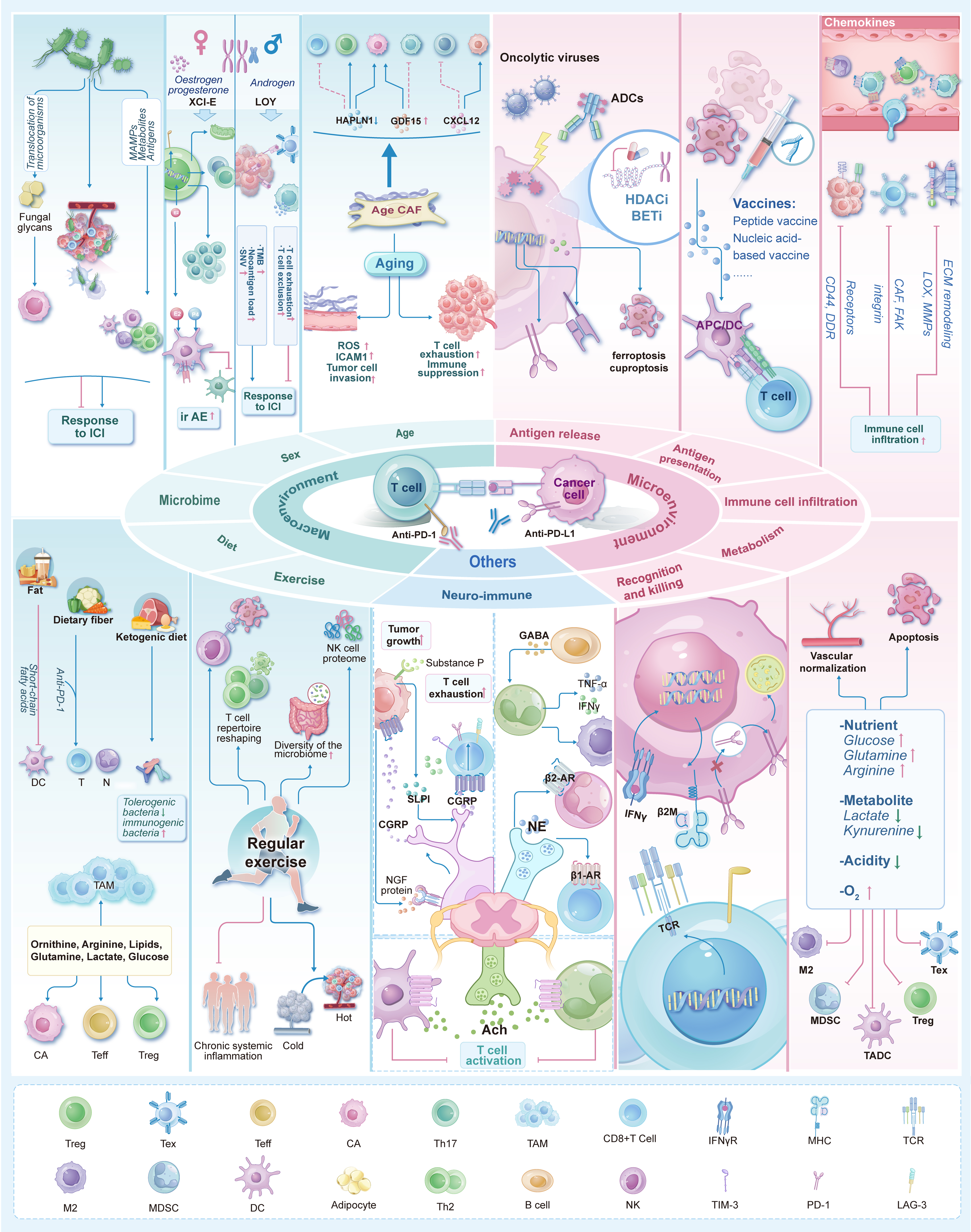
Figure 6. Potential factors influencing PD-1/PD-L1 inhibitor response. Multiple factors within both the macro- and microenvironments interact to regulate antitumor immunity and responses to PD-1/PD-L1 inhibitors. Microenvironmental factors involve elements affecting antigen release, antigen presentation, immune cell activation, and metabolism. Macroenvironmental factors include age, sex, microbiome, diet, and exercise. Recently, the influence of neuroimmune interactions on antitumor immunity has been increasingly recognized, suggesting additional potential targets for overcoming resistance. PD-1: Programmed death-1; PD-L1: programmed death-ligand 1; ADC: antibody-drug conjugate; HDACi: histone deacetylase inhibitor; BETi: bromodomain and extraterminal inhibitor; APC: antigen-presenting cell; DC: dendritic cell; ECM: extracellular matrix; LOX: lysyl oxidase; MMPs: matrix metalloproteinases; CAF: cancer-associated fibroblast; FAK: focal adhesion kinase; DDR: discoidin domain receptor; Tex: exhausted T cell; Treg: regulatory T cell; TADC: tumor-associated dendritic cell; MDSC: myeloid-derived suppressor cell; IFNγ: interferon gamma; β2M: beta-2 microglobulin; TCR: T cell receptor; GABA: gamma-aminobutyric acid; TNF-α: tumor necrosis factor alpha; β1-AR: beta-1 adrenergic receptor; β2-AR: beta-2 adrenergic receptor; NE: norepinephrine; SLPI: secretory leukocyte protease inhibitor; CGRP: calcitonin gene-related peptide; Ach: acetylcholine; NK: natural killer; CA: cancer cell; Teff: effector T cell; TAM: tumor-associated macrophage; N: neutrophil; MAMPs: microbe-associated molecular patterns; ICI: immune checkpoint inhibitor; XCI-E: X-chromosome inactivation escape; LOY: loss of Y chromosome; irAE: immune-related adverse event; TMB: tumor mutational burden; HAPLN1: hyaluronan and proteoglycan link protein 1; GDF15: growth differentiation factor 15; CXCL12: C-X-C motif chemokine ligand 12; ICAM1: intercellular adhesion molecule 1.
Promoting antigen release and presentation
The presence of sufficient neoantigens recognizable by the immune system is the first step in initiating antitumor immunity. Enhancing effective antigen release is a key strategy for improving the efficacy of anti-PD-1/PD-L1 inhibitors. Epigenetic drugs represent a classic approach to restoring tumor immunogenicity. However, early clinical trials revealed limited clinical activity when combining histone deacetylase (HDAC) inhibitors with PD-1/PD-L1 inhibitors[39]. In addition, novel epigenetic drugs, such as bromodomain and extraterminal (BET) inhibitors, have shown potential for enhancing the efficacy of anti-PD-1/PD-L1 therapies[40]. Oncolytic virus therapy is a novel antitumor strategy that enhances immunogenic cell death (ICD) in tumors. Mechanistic studies and early clinical trials have confirmed its role in sensitizing tumors to immunotherapy[41]. Recent research has focused on engineering oncolytic viruses that express multiple molecules, such as OX40L, to further enhance the effects of PD-1/PD-L1 inhibitors[42]. Antibody-drug conjugates (ADCs) carrying ICD-inducing payloads have also been shown to promote ICD, providing a rationale for combination strategies with immunotherapy. Several clinical studies have preliminarily demonstrated synergistic effects between ADCs and PD-1/PD-L1 inhibitors. Next-generation conjugates, such as bispecific ADCs, stimulator of interferon genes (STING) agonist-loaded ADCs, and conditionally active ADCs, hold promise for enhancing the efficacy of combination therapy while reducing treatment-related adverse events[43,44]. Additional methods to promote antigen release are currently being explored in preclinical studies. Antigen release often depends on ICD processes, such as apoptosis, necrosis, and regulated cell death (RCD), including ferroptosis, cuproptosis, pyroptosis, and programmed necrosis. However, most ICD occurs through apoptosis, which is generally immunotolerant and unable to activate antitumor immunity[45]. As a result, inducing RCD is emerging as a promising strategy for immunotherapy. Ferroptosis is a major focus in this field. In several mouse tumor models, ferroptosis inducers, such as GPX4 inhibitors and translocator protein (TSPO) inhibitors, have shown strong synergy with anti-PD-1/PD-L1 therapy[46,47]. Ferroptosis not only directly suppresses tumor cell proliferation, but also activates dendritic cells (DCs) by releasing damage-associated molecular patterns (DAMPs)[48]. Furthermore, induction of ferroptosis by targeting upstream or downstream molecules can also enhance immunotherapy efficacy, as demonstrated by TMOD3 knockdown or heterogeneous nuclear ribonucleoprotein L (HnRNP L) knockdown[49,50]. However, ferroptosis-induced immunogenicity is context-dependent[51]. Maximizing its therapeutic efficacy requires improving tumor selectivity while reducing the suppressive effects of oxidized lipids on immune cells[52,53]. Nanoparticles and similar materials represent a potential solution[54,55]. Cuproptosis, proposed in 2022, also shows promise for enhancing anti-PD-1/PD-L1 efficacy, but its clinical application still requires further preclinical research[56].
Tumor antigens are captured by antigen-presenting cells (APCs) and presented to T cells through major histocompatibility complex (MHC) molecules. Reduced numbers of DCs in draining lymph nodes and impaired T cell activation hinder antitumor immunity and limit the efficacy of PD-1/PD-L1 inhibitors. Clinical trials have evaluated antigen vaccines, CD40 agonists, and Toll-like receptor (TLR) agonists to improve responses to anti-PD-1/PD-L1 therapy, but the results have been mixed. A phase I trial showed that the TLR9 agonist vidutolimod plus pembrolizumab induced durable tumor regression in 25% of patients with metastatic melanoma resistant to PD-1 inhibitors[57], whereas a separate phase I study of a TLR7 agonist showed limited benefit either as monotherapy or in combination with a PD-1 inhibitor[58]. Preclinical studies are now exploring TLR agonists incorporated into nanomedicines or co-delivery systems to better reverse the suppressive tumor microenvironment (TME) and improve anti-PD-1/PD-L1 activity[59]. Neoantigen vaccines activate APCs by directly delivering antigens, and studies have shown that vaccine-induced memory T cells can provide long-term protection[60]. Personalized neoantigen vaccines combined with PD-1/PD-L1 blockade have demonstrated preliminary antitumor activity in early-phase trials[61]. Current preclinical studies aim to further optimize these combinations by refining neoantigen design, adjusting dosing schedules, and integrating multi-drug strategies. The functional state of DCs also shapes antigen presentation. Sánchez-Paulete et al. identified BATF3-dependent DCs as essential for cross-presenting tumor antigens and mediating anti-PD-1 responses[62]. The interaction between the STAT3 and STAT5 transcriptional pathways determines DC phenotypes in the TME, and STAT3 degraders represent a potential immunotherapeutic approach[63]. Another study found that TP63 and STAT1 mutually inhibit each other and regulate the IFN-γ signaling pathway through co-occupation and co-regulation of promoters and enhancers, and TP63 silencing can enhance PD-1 blockade efficacy[64].
Loss of co-stimulatory factors such as 4-1BB/4-1BBL and CD28/CD80 also impairs T cell activation and contributes to immunotherapy resistance. Combinations of 4-1BB agonists with PD-1/PD-L1 inhibitors, as well as bispecific antibodies, have entered clinical trials. Although the results have not been entirely satisfactory, high response rates have been observed in certain cancers (e.g., melanoma)[65]. CD28, another primary co-stimulatory receptor, provides the second activation signal by binding CD80/CD86. Despite early safety concerns, CD28 remains a viable therapeutic target. Majocchi et al. recently reported early results for NI-3201, a bispecific antibody that mediates PD-L1-dependent CD28 co-stimulation, and clinical studies are being planned[66]. However, another study has identified a distinct function of CD28 in tumor cells - promoting immune evasion. Knockdown of tumor-cell CD28 may overcome anti-PD-1 resistance by affecting PD-L1[67]. Because CD28 exerts different functions in tumor cells and T cells, therapeutic targeting requires strict cell specificity. RNA sequencing studies have also identified novel targets, such as mitochondrial antiviral signaling protein (MAVS) and FTSJ3, which modulate antitumor immunity through IFN signaling and may sensitize tumors to PD-1/PD-L1 blockade[68,69]. These findings offer new insights into combination strategies, though clinical translation remains ongoing.
Enhancing the trafficking and infiltration of immune cells
Modulating chemokine networks
Chemokines are key cytokines that regulate immune cell migration and lymphoid tissue development and are grouped into four families: C, CC, CXC, and CX3C. Immune cells show distinct migratory responses within the TME based on their chemokine receptor expression profiles. Antitumor immune cells, including CD8+ T cells, natural killer (NK) cells, and Th1 cells, mainly rely on CXCR3 and its ligands CXCL9/10, whereas regulatory T (Treg) cells depend on the CCR4-CCL22/28 axis, and myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) are driven by the CCR2-CCL2 axis[70]. Emerging evidence highlights the capacity of CXCL8 to increase macrophage PD-L1 levels by activating purine metabolism[71]. Therefore, modulating chemokines to reshape the immunosuppressive TME is a potential strategy to enhance anti-PD-1/PD-L1 activity. Early clinical trials combining CCR4 antagonists (e.g., mogamulizumab) or CXCR4 antagonists (e.g., BL-8040) with PD-1/PD-L1 inhibitors have demonstrated favorable safety profiles and preliminary antitumor activity[72,73]. Bergeron et al. reported that the addition of a CXCR2 antagonist to partial irradiation and anti-PD-1 therapy reduced irradiation-induced CXCR2-driven neutrophil infiltration and strengthened therapeutic efficacy[74]. Recent studies have also shown that regulating chemokine expression by targeting upstream regulators may overcome anti-PD-1/PD-L1 resistance. For instance, inhibition of lysine-specific demethylase 4C (KDM4C) increased CXCL10 transcription through enhanced H3K36me3 occupancy at the promoter region, and the combination of KDM4C inhibition, radiotherapy, and anti-PD-L1 treatment yielded the strongest antitumor activity with manageable toxicity in a lung cancer mouse model[75]. WNT11-overexpressing tumor cells limit CD8+ T cell recruitment by reducing CXCL10 and CCL4 through CAMKII-mediated β-catenin/AFF3 downregulation, and CAMKII inhibition has shown synergy with anti-PD-1 therapy in liver metastasis models[76]. Additional targets involved in chemokine signaling have also been identified, including translationally controlled tumor protein (TCTP), which activates the EGFR-AKT-MCL-1/CXCL10 pathway[77]; lymphotoxin beta receptor (LTβR), which induces tertiary lymphoid structure (TLS)-associated chemokines[78]; and zinc finger E-box-binding homeobox 1 (ZEB1), which suppresses CXCL10 secretion and restricts CD8+ T cell infiltration[79]. Although clinical trials focused on chemokine modulation remain limited, existing early clinical trials and preclinical evidence indicate that this strategy warrants continued investigation.
Normalizing tumor vasculature
When immune cells reach tumor sites, they undergo a complex process of infiltration and reactivation. Tumor cells promote tumor vascularization through vessel co-option and angiogenesis, which supports tumor growth and facilitates immune evasion. Immune cells in the TME can also stimulate angiogenesis. This bidirectional interaction between tumor vasculature and immune cells drives the development of an immunosuppressive phenotype. Anti-angiogenic therapy is one of the clinically validated strategies that enhance the activity of PD-1/PD-L1 inhibitors, but this regimen has not markedly improved OS in some cancers[80]. Tumor hypoxia is a major factor limiting therapeutic efficacy. Studies have shown that prolonged use of anti-angiogenic drugs at approved doses excessively prunes tumor vessels, worsening hypoxia and immune suppression, thereby reducing the effectiveness of immunotherapy[81]. Concurrent anti-angiogenic therapy may also reduce the intratumoral distribution of anti-PD-1/PD-L1 antibodies[82]. Additionally, the impact of vascular selectivity and “non-angiogenic” tumors is often underestimated. Tumors that rely on vessel co-option are inherently resistant to VEGF inhibitors and often exhibit poor immune cell infiltration[83]. Future strategies should move beyond simple vascular blockade toward more precise regulation. Sustaining a prolonged “vascular normalization” window by lowering drug doses and shortening treatment cycles has been shown to improve perfusion and immune cell infiltration[84]. Co-inhibition of VEGF and hypoxia-inducible factor-1α (HIF-1α) within the hypoxia pathway promotes vascular normalization and strengthens PD-1/PD-L1 blockade efficacy[85]. In addition, multi-target combinations and novel delivery platforms represent emerging approaches to normalizing tumor vasculature and overcoming resistance to PD-1/PD-L1 inhibitors[86,87].
Remodeling the extracellular matrix
The extracellular matrix (ECM) is a crucial component of the TME. It serves as a physical barrier that impedes immune cell infiltration and regulates immune cell activity through biochemical signaling. The dual physical and physiological barriers created by ECM remodeling are considered a mechanism of immunotherapy resistance. Cancer-associated fibroblasts (CAFs) are drivers of ECM remodeling and secrete several collagens and cross-linking enzymes to construct a dense fibrotic matrix. Enhancing active immune cell infiltration by targeting CAFs has proven to be an effective approach to potentiating PD-1/PD-L1 blockade[88,89]. Lysyl oxidase (LOX) catalyzes collagen cross-linking, thereby increasing matrix stiffness and activating pro-cancer signaling pathways, such as the integrin-FAK-Src pathway[90]. In addition, this process increases interstitial pressure, thereby restricting substance transport. Matrix metalloproteinases (MMPs), secreted by CAFs and TAMs, degrade the ECM while recruiting MDSCs and activating CAFs[91]. Discoidin domain receptors (DDRs), which are transmembrane tyrosine kinase receptors, enhance collagen remodeling and thereby promote immune exclusion[92]. CD44, the primary receptor for hyaluronic acid (HA), promotes macrophage M2 polarization and Treg recruitment upon HA binding[93]. Disrupting CD44-HA interactions to target TAMs may improve immunotherapy efficacy. Although breakthroughs have been made in developing drugs targeting pathways involving LOX, MMPs, DDRs, and CD44, multi-targeted therapy is a more rational option because of the complexity of ECM-TME interactions. Research on pancreatic cancer has shown that the combined targeting of intracellular and ECM components enhances the efficacy of anti-PD-1/PD-L1 therapy[94]. Furthermore, Ishihara et al. demonstrated that conjugating anti-PD-L1 antibodies with ECM super-affinity peptides increases drug accumulation in tumor tissues while decreasing systemic concentrations, thereby enhancing efficacy and reducing adverse reactions[95]. Photodynamic therapy using PD-L1 immune checkpoint-targeted photoactivable liposomes (iTPALs) has also been shown to block PD-1/PD-L1 more effectively than free anti-PD-1/PD-L1 antibodies by reducing collagen density[96]. These preclinical findings highlight the potential of targeting the ECM to improve the efficacy of PD-1/PD-L1 inhibitors and support the development of related combination strategies for future clinical trials.
Targeting immune cell function
The functional state and interactions of immune cells in the TME are critical factors influencing antitumor immunity. Studies have confirmed that increasing the effector T cell/Treg ratio by targeting CD25+ Treg cells enhances the efficacy of anti-PD-1/PD-L1 therapy[97]. Another study found that Src family kinase inhibitors improve the efficacy of PD-1/PD-L1 blockade by inhibiting Treg conversion and proliferation[98]. Single-cell sequencing has identified neutrophil subsets associated with PD-1 inhibitor resistance, which suppress antitumor immunity by inducing irreversible T cell exhaustion[99]. Macrophages are another important component of the TME and are categorized as classically activated M1 and alternatively activated M2 phenotypes. M1 macrophages primarily exert proinflammatory and antitumor effects, whereas M2 macrophages exhibit immunosuppressive and protumor functions. However, macrophages in the TME are predominantly M2-like TAMs, which promote Treg differentiation and suppress effector T cell function[100]. TAM enrichment has been associated with resistance to ICIs, regardless of PD-L1 expression, suggesting that modulating TAMs could enhance immunotherapy[101]. Notably, multi-omics analyses have revealed elevated levels of M2 macrophages in patients who do not respond to anti-PD-1/PD-L1 combined with anti-angiogenic therapy[102]. Selective inhibition of molecules associated with M2 macrophage polarization can reprogram macrophages and overcome PD-L1 inhibitor resistance. Additionally, direct intratumoral delivery of mRNA encoding a membrane-anchored anti-CD3 single-chain variable fragment (scFv) can reprogram TAMs and promote activation of tumor-infiltrating lymphocytes. In tumor models, silencing triggering receptor expressed on myeloid cells 1 (TREM1) significantly prolonged survival. Further mechanistic research demonstrated that TREM1 deficiency enhances anti-PD-1 activity by limiting MDSC function and preventing T cell exhaustion[103]. Prasad et al. reported that early treatment with trametinib reduces MDSC abundance by suppressing CSF-1 expression, thereby sensitizing tumors to anti-PD-1 therapy, but long-term treatment restores CSF-1 expression and abolishes anti-PD-1 activity[104]. However, a phase I trial combining the CSF-1R inhibitor pexidartinib with durvalumab for advanced cancers showed limited clinical activity. Further analysis suggested that this may be related to the adverse effects of pexidartinib on DC differentiation through FLT3 inhibition[105]. Collectively, these findings suggest that targeting a single immune cell subset is inadequate for bypassing anti-PD-1/PD-L1 resistance, highlighting the necessity of multifaceted interventions to reverse the immunosuppressive TME.
Regulating metabolism in the TME
Abnormal metabolism contributes to an immunosuppressive TME. Enhancing the therapeutic efficacy of PD-1/PD-L1 blockade through metabolic reprogramming is an emerging and promising approach. In a preclinical study of colorectal cancer, H2S promoted Treg activation and inhibited CD8+ T cell migration via ENO1 and ELK4 post-sulfation. Conversely, reducing H2S can enhance the antitumor activity of ICIs[106]. Boelaars et al. reported that sialic acid deficiency increases CD4+ and CD8+ T cells while reducing CD4+ Tregs, thereby sensitizing pancreatic cancer cells to immunotherapy[107]. Elevated blood ammonia levels in colorectal cancer patients were associated with non-response to anti-PD-L1 therapy[108]. Another study indicated that supplying L-arginine to immune cells while limiting its uptake by tumor cells may be an optimal strategy[109]. Dynamic labeling of L-arginine using nanomaterials can improve its delivery and boost ICI efficacy[110]. Other research also showed that inhibiting glutamine metabolism in tumor cells enhances immune cell function and synergizes with PD-L1 blockade[111]. Hypoxia in the TME is another factor contributing to anti-PD-1/PD-L1 resistance. Alleviating hypoxia through direct or indirect oxygen delivery has significantly improved antitumor efficacy. Researchers developed alginate microcapsules encapsulating cyanobacteria that generate sustained oxygen via photosynthesis. In a breast cancer mouse model, this approach demonstrated strong synergy with anti-PD-1 therapy[112]. A recent study proposed that targeting lipid rafts and mitochondrial respiration with albumin-bound statins can reduce hypoxia and improve PD-1 blockade efficacy in NSCLC[113].
Promoting T cell recognition and killing of tumor cells
Activated T cells recognize tumor cells through T cell receptor (TCR)-MHC-peptide interactions and eliminate them with the support of co-stimulatory molecules. Studies have shown that increasing MHC levels through PCSK9 inhibition, α-ketoglutarate supplementation, or G3BP1 inhibition can enhance T cell recognition of tumor cells[114-116]. Recent research revealed that valosin-containing protein (VCP) stabilizes GPD1L, causing downstream glycerol-3-phosphate (G3P) accumulation and subsequent TCR impairment. Dual inhibition of VCP and PD-1 enhanced antitumor activity in a mouse model of HCC[117]. The integration of PD-1/PD-L1 blockade and adoptive cell therapy (ACT) has also been actively investigated. Physical and immunologic barriers suppress ACT efficacy within solid malignancies, and the addition of PD-1/PD-L1 inhibitors could help overcome these challenges[118]. Giuffrida et al. reported that interleukin (IL)-7/IL-15 pretreatment improves chimeric antigen receptor T cell (CAR-T) responses to anti-PD-1/PD-L1 therapy[119]. Advances in the ex vivo expansion and delivery of specific effector T cells may further strengthen this combination therapy. Simultaneous blockade of PD-1 and other immune checkpoints has shown favorable safety profiles and antitumor activity in multiple phase III trials. However, limited efficacy and safety concerns in certain cancers persist, highlighting the need for further mechanistic studies[120]. A preclinical study suggested that a TIGIT and 4-1BB bispecific antibody (ABL112), when combined with pembrolizumab, inhibits tumor growth more effectively than pembrolizumab plus an anti-TIGIT antibody[121]. Hu et al. found that immunoradiotherapy with dual LAG-3 and TIGIT blockade effectively overcomes anti-PD-1 resistance[122]. Targeting multiple immune checkpoints remains a promising strategy to strengthen PD-1/PD-L1 inhibitors, and advances in multi-target inhibitors and delivery technologies may further improve clinical responses.
Modulating host factors
Beyond the microenvironment, host factors such as age, sex, microbiota, and diet also influence the therapeutic outcomes of PD-1/PD-L1 blockade. With increasing age, lymphoid tissues undergo structural and cellular alterations that lead to diminished immune surveillance and a peripheral pro-inflammatory immune environment. This decline in immune adaptability is not only associated with increased cancer risk but also impacts the efficacy of immunotherapy[123]. Although immune dysfunction worsens with age across most tumor types, the roles of specific immune cell subsets are cancer type-specific. Furthermore, aging also reshapes the immune microenvironment through alterations in the ECM, blood vessels, adipocytes, and other components.
A meta-analysis showed that males may benefit more from immunotherapy, but another meta-analysis in NSCLC reported the opposite finding. Sex-related differences arise from complex interactions among genetic, hormonal, behavioral, and environmental factors. Among these, sex hormones - particularly estrogen and androgen - are key regulators of antitumor immunity and responses to ICIs. Estrogen and its receptors (ERα and ERβ) influence the formation of the TME through both tumor cell-intrinsic and -extrinsic mechanisms. Studies in NSCLC have shown that tumor cells produce estrogen through aromatase expression and activate ERα via an autocrine mechanism, thereby promoting CD274/PD-L1 overexpression[124]. Another preclinical study indicated that estrogen promotes the recruitment of TAMs and induces their polarization toward the M2 phenotype[125]. Adurthi et al. found that ERα signaling stimulates Treg infiltration by directly binding to the FOXP3 promoter and upregulating the expression of immunosuppressive molecules[126]. Furthermore, in melanoma, lung cancer, and ovarian cancer models, estrogen was shown to promote the immunosuppressive activity of MDSCs and their infiltration into tumor sites by activating the JAK2-STAT3 or TNFα pathways[127,128]. Preclinical models have demonstrated that blocking estrogen signaling with selective ER-degrading agents effectively restores CD8+ T cell function and enhances the efficacy of ICIs[125,129]. Notably, estrogen-mediated regulation of T cell function exhibits complexity that is dependent on receptor subtype. Binding to ERα restricts the proliferation and intratumoral infiltration of cytotoxic T cells by inducing mitochondrial dysfunction, shortening telomere length, and suppressing hTERT expression[130,131]. However, activation of ERβ in CD8+ T cells enhances TCR signaling by modulating tumor-derived phosphotyrosine switches, thereby increasing anti-PD-1 activity[132]. The effects of androgens in the TME primarily involve driving CD8+ T cell exhaustion and suppressing innate immune cell function. The androgen receptor (AR) directly impairs CD8+ T cell function by binding to androgen response elements (AREs) within open chromatin regions of the IFN-γ and GZMB genes, thereby suppressing their expression[133]. Studies in bladder cancer models have also found that AR promotes the early differentiation of CD8+ T cells into a “progenitor-exhausted” phenotype by upregulating the transcription factors TCF7/TCF1[134]. Furthermore, Liu et al. found that anti-androgen therapy or AR knockdown can reduce PD-L1 expression in NK cells by downregulating circ_0001005[135]. Based on these mechanisms, blocking androgen signaling via androgen deprivation therapy (ADT) or AR antagonists has been shown to exert synergistic effects with anti-PD-1/PD-L1 therapy in several preclinical models[133,136]. In addition to estrogen and androgen, progesterone and human chorionic gonadotropin (hCG) have also been found to influence T cell epigenetic and metabolic reprogramming by upregulating progesterone-induced inhibitory factors and promoting histone methylation[9]. Recent studies have further revealed sex-specific differences in the function of certain enzymes within T cells. In female mice, the absence of diacylglycerol acyltransferase 1 (DGAT1) in T cells enhances antitumor immunity by improving mitochondrial function and expanding precursor-depleted T cells. However, in male mice, DGAT1 deficiency leads to fatty acid peroxidation, endoplasmic reticulum stress, and T cell death via AR signaling, thereby impairing antitumor immunity[137]. The mechanisms underlying sex-related differences in tumor immunity and immunotherapy responses are complex. Current studies suggest that modulating sex hormones to enhance sensitivity to anti-PD-1/PD-L1 therapy is a promising direction, but further mechanistic studies are needed.
Long-distance regulation by gut microbiota and local remodeling via intratumoral microbiota together influence the TME. Gut microbiota not only contribute to local tumorigenesis but also influence tumor immunity through long-distance regulation of the TME[138]. Studies in mice have revealed differential effects of the gut microbiota on patient responses to anti-PD-1/PD-L1 treatment[139]. Fidelle et al. found that antibiotic-induced dysbiosis increases intestinal Enterocloster abundance and causes a loss of MAdCAM-1 expression, thereby triggering immune-suppressive Treg17 cell infiltration into the TME[140]. Another study showed that specific gut microbiota can downregulate PD-L2 and its ligand RGMb, and that blocking this pathway may overcome anti-PD-1 resistance[141]. Ex-Th17 cells induced by Enterobacteriaceae were found to produce high levels of IFN-γ and TNF in the TME, thereby promoting antitumor immunity and achieving anti-PD-1-mediated tumor control[142]. Furthermore, soluble metabolites from gut bacteria, such as short-chain fatty acids, inosine, and bile acids, can reach the TME via systemic circulation and influence tumor immunity[138]. Additional clinical evidence suggests that the use of broad-spectrum antibiotics disrupts the gut microbiome, thereby impairing immunotherapy efficacy[143]. Conversely, increased gut microbiota diversity (e.g., Bifidobacterium spp.) has been associated with higher response rates to ICIs[139]. Modulation of the gut microbiota via fecal microbiota transplantation has also been shown to enhance the efficacy of PD-1 inhibitors in early clinical trials[144]. Recently, a synthetic microbiota composed of 15 gut bacterial strains was successfully engineered and demonstrated the potential to reverse anti-PD-1 antibody resistance, regardless of individual gut microbiota backgrounds[145]. Unlike gut microbiota, microorganisms colonizing tumor sites directly participate in TME remodeling. Although the biomass of intratumoral bacteria is low (typically 1-10 bacteria per 100,000 tumor cells)[146], these microorganisms can influence the TME through direct contact and local metabolism. H. pylori and S. Typhimurium promote immune evasion by inducing PD-L1 expression in host cells[147,148]. Hezaveh et al. found that indole metabolites secreted by Lactobacillus promote the polarization of TAMs toward an immunosuppressive phenotype by activating the AhR pathway[149]. Conversely, some bacteria are considered beneficial for immunotherapy, with baseline enrichment of Clostridium at tumor sites found to be associated with response to ICIs[150]. Intratumoral injection of Burkholderia cepacia, Priestia megaterium, or Corynebacterium kroppenstedtii in melanoma-bearing mice was found to synergistically inhibit tumor growth when combined with anti-PD-1 therapy[151]. With the deepening understanding of the microbiome, clinical intervention strategies are shifting toward precision medicine. Canale et al. found that engineered tumor-colonizing Escherichia coli can convert ammonia into L-arginine, thereby improving immunotherapy efficacy[152].
In addition to the factors discussed above, diet, exercise, and chronic stress may also influence tumor immunity and responses to anti-PD-1/PD-L1 therapy. Preclinical studies have shown that a ketogenic diet alters the immunosuppressive TME through multiple mechanisms, increasing sensitivity to ICIs[153]. Regular exercise has been shown to enhance NK cell cytotoxicity and reshape the T cell repertoire, supporting antitumor immunity[154]. Growing interest in host-related factors underscores the potential of modulating host physiology to improve immunotherapy efficacy.
Targeting neuro-immune crosstalk
With increasing research attention focused on the neuro-immune system, its role in ICI resistance has gradually been recognized in recent years. Abnormal nerve fiber growth or irregular neurotransmitter secretion can lead to immune escape. Targeting neurotransmitters and their receptors has shown potential for overcoming resistance. Plasma neurotransmitter analyses in patients who were sensitive or resistant to anti-PD-1 antibodies revealed that norepinephrine influences CXCL9 and adenosine (ADO) secretion in tumor cells, thereby inhibiting the chemotaxis and function of CD8+ T cells[155]. Qiao et al. reported that norepinephrine inhibits TCR signaling by activating β-adrenergic receptors (β-ARs) on CD8+ T cells while promoting metabolic dysfunction and exhaustion[156]. A meta-analysis showed that β-blocker use may correlate with improved ICI efficacy[157]. The safety and antitumor activity of combining β-AR antagonists with PD-1/PD-L1 inhibitors are being investigated in several clinical trials. In contrast to the β-AR pathway, α2-adrenergic receptor (α2-AR) agonists have shown the ability to induce MDSC apoptosis and enhance CD8+ T cell infiltration, thereby exhibiting effective antitumor activity even in ICI-resistant tumor models[158]. Furthermore, activation of the α7 nicotinic acetylcholine receptor has been shown to suppress TNF-α release in macrophages and modulate T cell function[159]. Schneider et al. revealed that platelet-derived serotonin upregulates PD-L1 on cancer cells, thereby driving CD8+ T cell exhaustion[160]. Combining serotonin-targeting drugs with PD-1/PD-L1 inhibitors has achieved long-term tumor inhibition. Further studies have also revealed the role of sensory neurons in tumor immunity. In preclinical melanoma models, calcitonin gene-related peptide (CGRP), released by nociceptors, increased CD8+ T cell exhaustion[161]. Although the mechanism by which CGRP regulates T cells remains incompletely understood, it may involve Gαs signaling mediated by the RAMP1–CALCRL receptor complex[162]. In summary, neurotransmitter signaling pathways play critical roles in resistance to PD-1/PD-L1 inhibitors. Further studies in this field may reveal novel therapeutic targets and combination strategies for overcoming immunotherapy resistance.
CONCLUSION AND FUTURE PERSPECTIVES
Recent years have seen PD-1/PD-L1 inhibitors revolutionize the landscape of antitumor therapy, but primary and acquired resistance remain substantial clinical challenges. Based on analyses of clinical and preclinical evidence, this review supports the view that mechanism-driven combination therapy is an inevitable trend toward achieving durable and effective immunotherapy responses. The success of OPDUALAG and ivonescimab has highlighted the clinical value of dual-target regimens, further driving the development of dual- or multi-target drugs. However, some combination regimens, such as anti-PD-L1 plus anti-TGF-β, have shown inconsistent efficacy or additive toxicity in clinical trials. These findings suggest that not all combinations are viable, and that combination strategies should be based on interpretable synergistic efficacy and manageable safety. One direction for future research is the development of conditionally active antibodies, ADCs, and nanomedicine-based delivery systems that restrict immune activation to the TME, thereby maximizing efficacy while minimizing systemic toxicity.
Furthermore, the mechanisms of resistance to PD-1/PD-L1 inhibitors exhibit high heterogeneity, suggesting that precision-tailored treatment regimens for tumors with different immune phenotypes may more effectively overcome resistance. For tumors characterized by dense stroma and immune exclusion, such as pancreatic cancer, improving immune cell infiltration by targeting the ECM and CAFs may improve the efficacy of anti-PD-1/PD-L1 therapy. Conversely, for tumors characterized by immune cell exhaustion, restoring T cell cytotoxicity through pathways such as metabolic reprogramming holds greater potential for achieving synergistic effects.
Notably, the influence of host factors on immunotherapy should be considered in future preclinical and clinical studies. Early trials have already recognized the value of modulating the gut microbiota and implementing sex hormone-based interventions to improve immunotherapy outcomes, marking a transition within the therapeutic paradigm from a tumor-centric to a host-centric approach. The next breakthrough in cancer immunotherapy will rely on dynamic, multi-omics-guided analyses to design personalized, multi-target treatment regimens. By comprehensively considering the intrinsic mechanisms of tumors, the TME, and host factors, the ultimate goal is to convert non-responders into responders, thereby achieving durable treatment responses in a broader patient population.
DECLARATIONS
Authors’ contributions
Conceptualization, methodology, writing of the original draft: Li L, Zhao Y
Data curation, formal analysis, visualization: Shi X, Guo X
Investigation, resources: Zhang X, Li G
Supervision, project administration, writing - review and editing: Meng Q, Zhang M, Yin M
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Chongqing Science & Health Joint Medical Research Program (Grant No. 2026MSXM148), the Chongqing Municipal Education Commission Science and Technology Research Program (Grant No. KJQN202500112), the Chongqing Young and Middle-aged Medical High-level Talents Program (Grant No. YXGD202544), and the Chongqing Key Special Project for Technological Innovation and Application Development (Grant No. CSTB2024TIAD-KPX0050).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887-95.
2. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141-51.
3. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365-9.
4. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-34.
5. Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793-800.
6. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293-7.
7. Curiel TJ, Wei S, Dong H, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562-7.
8. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2022;185:576.
9. Conforti F, Pala L, Di Mitri D, et al. Sex hormones, the anticancer immune response, and therapeutic opportunities. Cancer Cell. 2025;43:343-60.
10. Cortellini A, Bersanelli M, Buti S, et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: when overweight becomes favorable. J Immunother Cancer. 2019;7:57.
11. Blake SJ, Wolf Y, Boursi B, Lynn DJ. Role of the microbiota in response to and recovery from cancer therapy. Nat Rev Immunol. 2024;24:308-25.
16. KEYTRUDA® (pembrolizumab) injection, for intravenous use. 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/125514s172lbl.pdf. [Last accessed on 11 May 2026].
17. TECENTRIQ® (atezolizumab) injection, for intravenous use. 2016. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/761034s053lbl.pdf. [Last accessed on 11 May 2026].
18. Zhao S, Zhao H, Yang W, Zhang L. The next generation of immunotherapies for lung cancers. Nat Rev Clin Oncol. 2025;22:592-616.
19. Laktionov K, Smolin A, Stroyakovskiy D, et al. Prolgolimab with chemotherapy as first-line treatment for advanced non-squamous non-small-cell lung cancer. Eur J Cancer. 2025;217:115255.
20. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
21. Mao Y, Xie H, Lv M, et al. The landscape of objective response rate of anti-PD-1/L1 monotherapy across 31 types of cancer: a system review and novel biomarker investigating. Cancer Immunol Immunother. 2023;72:2483-98.
22. Lang R, Welponer T, Richtig E, et al. Nivolumab for locally advanced and metastatic cutaneous squamous cell carcinoma (NIVOSQUACS study)-Phase II data covering impact of concomitant haematological malignancies. J Eur Acad Dermatol Venereol. 2023;37:1799-810.
23. Kaufman HL, Russell JS, Hamid O, et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J Immunother Cancer. 2018;6:7.
24. Yau T, Park JW, Finn RS, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022;23:77-90.
25. Klein C, Brinkmann U, Reichert JM, Kontermann RE. The present and future of bispecific antibodies for cancer therapy. Nat Rev Drug Discov. 2024;23:301-19.
26. Finn RS, Qin S, Ikeda M, et al.; IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894-905.
27. Reck M, Mok TSK, Nishio M, et al.; IMpower150 Study Group. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. 2019;7:387-401.
28. Douma LAH, van der Noort V, Lalezari F, et al. Pembrolizumab plus lenvatinib as second-line treatment in patients with pleural mesothelioma (PEMMELA): cohort 2 of a single-arm, phase 2 study. Lancet Oncol. 2025;26:1676-84.
29. Motzer RJ, Jonasch E, Agarwal N, et al. NCCN Guidelines® Insights: kidney cancer, version 2.2024. J Natl Compr Canc Netw. 2024;22:4-16.
30. Xiong A, Wang L, Chen J, et al. Ivonescimab versus pembrolizumab for PD-L1-positive non-small cell lung cancer (HARMONi-2): a randomised, double-blind, phase 3 study in China. Lancet. 2025;405:839-49.
31. Chen Z, Yang F, Jiang Z, et al. Ivonescimab plus chemotherapy versus tislelizumab plus chemotherapy as first-line treatment for advanced squamous non-small-cell lung cancer (HARMONi-6): a randomised, double-blind, phase 3 trial. Lancet. 2025;406:2078-88.
32. Johnson ML, Cho BC, Luft A, et al.; POSEIDON investigators. Durvalumab with or without tremelimumab in combination with chemotherapy as first-line therapy for metastatic non-small-cell lung cancer: the phase III POSEIDON study. J Clin Oncol. 2023;41:1213-27.
33. Paz-Ares L, Ciuleanu TE, Cobo M, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:198-211.
34. Tawbi HA, Schadendorf D, Lipson EJ, et al.; RELATIVITY-047 Investigators. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386:24-34.
35. Merck KGaA, Darmstadt, Germany Statement on phase II study of Bintrafusp Alfa in first-line treatment of biliary tract cancer. Available from: https://www.emdgroup.com/en/news/bintrafusp-alfa-update-23-08-2021.html. [Last accessed on 11 May 2026].
36. Cho BC, Lee JS, Wu YL, et al. Bintrafusp Alfa versus pembrolizumab in patients with treatment-naive, programmed death-ligand 1-high advanced NSCLC: a randomized, open-label, phase 3 trial. J Thorac Oncol. 2023;18:1731-42.
37. Masuda J, Sakai H, Tsurutani J, et al. Efficacy, safety, and biomarker analysis of nivolumab in combination with abemaciclib plus endocrine therapy in patients with HR-positive HER2-negative metastatic breast cancer: a phase II study (WJOG11418B NEWFLAME trial). J Immunother Cancer. 2023;11:e007126.
38. Spigel D, Jotte R, Nemunaitis J, et al. Randomized phase 2 studies of checkpoint inhibitors alone or in combination with pegilodecakin in patients with metastatic NSCLC (CYPRESS 1 and CYPRESS 2). J Thorac Oncol. 2021;16:327-33.
39. Levy BP, Giaccone G, Besse B, et al. Randomised phase 2 study of pembrolizumab plus CC-486 versus pembrolizumab plus placebo in patients with previously treated advanced non-small cell lung cancer. Eur J Cancer. 2019;108:120-8.
40. Wang ZQ, Zhang ZC, Wu YY, et al. Bromodomain and extraterminal (BET) proteins: biological functions, diseases, and targeted therapy. Signal Transduct Target Ther. 2023;8:420.
41. Nassiri F, Patil V, Yefet LS, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med. 2023;29:1370-8.
42. Liu S, Li F, Ma Q, et al. OX40L-armed oncolytic virus boosts T-cell response and remodels tumor microenvironment for pancreatic cancer treatment. Theranostics. 2023;13:4016-29.
43. Zhang P, Rashidi A, Zhao J, et al. STING agonist-loaded, CD47/PD-L1-targeting nanoparticles potentiate antitumor immunity and radiotherapy for glioblastoma. Nat Commun. 2023;14:1610.
44. Tsuchikama K, Anami Y, Ha SYY, Yamazaki CM. Exploring the next generation of antibody-drug conjugates. Nat Rev Clin Oncol. 2024;21:203-23.
45. Rongvaux A, Jackson R, Harman CC, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563-77.
46. Conche C, Finkelmeier F, Pešić M, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72:1774-82.
47. Tao Q, Liu N, Wu J, Chen J, Chen X, Peng C. Mefloquine enhances the efficacy of anti-PD-1 immunotherapy via IFN-γ-STAT1-IRF1-LPCAT3-induced ferroptosis in tumors. J Immunother Cancer. 2024;12:e008554.
48. Gong D, Chen M, Wang Y, Shi J, Hou Y. Role of ferroptosis on tumor progression and immunotherapy. Cell Death Discov. 2022;8:427.
49. He Z, Zheng D, Li F, et al. TMOD3 accelerated resistance to immunotherapy in KRAS-mutated pancreatic cancer through promoting autophagy-dependent degradation of ASCL4. Drug Resist Updat. 2025;78:101171.
50. Zhou X, Zou L, Liao H, et al. Abrogation of HnRNP L enhances anti-PD-1 therapy efficacy via diminishing PD-L1 and promoting CD8+ T cell-mediated ferroptosis in castration-resistant prostate cancer. Acta Pharm Sin B. 2022;12:692-707.
51. Catanzaro E, Demuynck R, Naessens F, Galluzzi L, Krysko DV. Immunogenicity of ferroptosis in cancer: a matter of context? Trends Cancer. 2024;10:407-16.
52. Wiernicki B, Maschalidi S, Pinney J, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13:3676.
53. Ma X, Xiao L, Liu L, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001-12.e5.
54. Gao Z, Zhang J, Hou Y, et al. Boosting the synergism between cancer ferroptosis and immunotherapy via targeted stimuli-responsive liposomes. Biomaterials. 2024;305:122442.
55. Wu B, Yang X, Kong N, Liang J, Li S, Wang H. Engineering modular peptide nanoparticles for ferroptosis-enhanced tumor immunotherapy. Angew Chem Int Ed Engl. 2025;64:e202421703.
56. Yan C, Liu Y, Zhao G, et al. Inhalable metal-organic framework-mediated cuproptosis combined with PD-L1 checkpoint blockade for lung metastasis synergistic immunotherapy. Acta Pharm Sin B. 2024;14:2281-97.
57. Ribas A, Medina T, Kirkwood JM, et al. Overcoming PD-1 blockade resistance with CpG-A Toll-like receptor 9 agonist vidutolimod in patients with metastatic melanoma. Cancer Discov. 2021;11:2998-3007.
58. Curigliano G, Jimenez MM, Shimizu T, et al. A phase I trial of LHC165 single agent and in combination with spartalizumab in patients with advanced solid malignancies. ESMO Open. 2024;9:103643.
59. Yu X, Long Y, Chen B, et al. PD-L1/TLR7 dual-targeting nanobody-drug conjugate mediates potent tumor regression via elevating tumor immunogenicity in a host-expressed PD-L1 bias-dependent way. J Immunother Cancer. 2022;10:e004590.
60. Braun DA, Moranzoni G, Chea V, et al. A neoantigen vaccine generates antitumour immunity in renal cell carcinoma. Nature. 2025;639:474-82.
61. Weber JS, Carlino MS, Khattak A, et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomised, phase 2b study. Lancet. 2024;403:632-44.
62. Sánchez-Paulete AR, Cueto FJ, Martínez-López M, et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 2016;6:71-9.
63. Zhou J, Tison K, Zhou H, et al. STAT5 and STAT3 balance shapes dendritic cell function and tumour immunity. Nature. 2025;643:519-28.
64. Jiang Y, Zheng Y, Zhang YW, et al. Reciprocal inhibition between TP63 and STAT1 regulates anti-tumor immune response through interferon-γ signaling in squamous cancer. Nat Commun. 2024;15:2484.
65. Khushalani NI, Ott PA, Ferris RL, et al. Final results of urelumab, an anti-CD137 agonist monoclonal antibody, in combination with cetuximab or nivolumab in patients with advanced solid tumors. J Immunother Cancer. 2024;12:e007364.
66. Majocchi S, Lloveras P, Nouveau L, et al. NI-3201 is a bispecific antibody mediating PD-L1-dependent CD28 co-stimulation on T cells for enhanced tumor control. Cancer Immunol Res. 2025;13:365-83.
67. Yang Z, Liu X, Zhu J, et al. Inhibiting intracellular CD28 in cancer cells enhances antitumor immunity and overcomes anti-PD-1 resistance via targeting PD-L1. Cancer Cell. 2025;43:86-102.e10.
68. Hwang BJ, Tsao LC, Acharya CR, et al. Sensitizing immune unresponsive colorectal cancers to immune checkpoint inhibitors through MAVS overexpression. J Immunother Cancer. 2022;10:e003721.
69. Zhuang Q, Dai Z, Xu X, et al. RNA methyltransferase FTSJ3 regulates the type I interferon pathway to promote hepatocellular carcinoma immune evasion. Cancer Res. 2024;84:405-18.
70. Bule P, Aguiar SI, Aires-Da-Silva F, Dias JNR. Chemokine-directed tumor microenvironment modulation in cancer immunotherapy. Int J Mol Sci. 2021;22:9804.
71. Yang L, Li A, Yu W, et al. Blockade of purine metabolism reverses macrophage immunosuppression and enhances anti-tumor immunity in non-small cell lung cancer. Drug Resist Updat. 2025;78:101175.
72. Doi T, Muro K, Ishii H, et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res. 2019;25:6614-22.
73. Bockorny B, Semenisty V, Macarulla T, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26:878-85.
74. Bergeron P, Dos Santos M, Sitterle L, et al. Non-homogenous intratumor ionizing radiation doses synergize with PD1 and CXCR2 blockade. Nat Commun. 2024;15:8845.
75. Jie X, Chen Y, Zhao Y, et al. Targeting KDM4C enhances CD8+ T cell mediated antitumor immunity by activating chemokine CXCL10 transcription in lung cancer. J Immunother Cancer. 2022;10:e003716.
76. Jiang W, Guan B, Sun H, et al. WNT11 promotes immune evasion and resistance to anti-PD-1 therapy in liver metastasis. Nat Commun. 2025;16:1429.
77. Lee HJ, Song KH, Oh SJ, et al. Targeting TCTP sensitizes tumor to T cell-mediated therapy by reversing immune-refractory phenotypes. Nat Commun. 2022;13:2127.
78. An D, Chen G, Cheng WY, et al. LTβR agonism promotes antitumor immune responses via modulation of the tumor microenvironment. Cancer Res. 2024;84:3984-4001.
79. Plaschka M, Benboubker V, Grimont M, et al. ZEB1 transcription factor promotes immune escape in melanoma. J Immunother Cancer. 2022;10:e003484.
80. Kuo HY, Khan KA, Kerbel RS. Antiangiogenic-immune-checkpoint inhibitor combinations: lessons from phase III clinical trials. Nat Rev Clin Oncol. 2024;21:468-82.
81. Rapisarda A, Melillo G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat Rev Clin Oncol. 2012;9:378-90.
82. Arjaans M, Schröder CP, Oosting SF, Dafni U, Kleibeuker JE, de Vries EG. VEGF pathway targeting agents, vessel normalization and tumor drug uptake: from bench to bedside. Oncotarget. 2016;7:21247-58.
83. Guelfi S, Hodivala-Dilke K, Bergers G. Targeting the tumour vasculature: from vessel destruction to promotion. Nat Rev Cancer. 2024;24:655-75.
84. Huang Y, Yuan J, Righi E, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561-6.
85. Lan J, Zeng R, Li Z, et al. Biomimetic nanomodulators with synergism of photothermal therapy and vessel normalization for boosting potent anticancer immunity. Adv Mater. 2024;36:e2408511.
86. Schmittnaegel M, Rigamonti N, Kadioglu E, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. 2017;9:eaak9670.
87. Liu Y, Zhan Z, Kang Z, et al. Preclinical and early clinical studies of a novel compound SYHA1813 that efficiently crosses the blood-brain barrier and exhibits potent activity against glioblastoma. Acta Pharm Sin B. 2023;13:4748-64.
88. Zhang Y, Fang Z, Pan D, et al. Dendritic polymer-based nanomedicines remodel the tumor stroma: improve drug penetration and enhance antitumor immune response. Adv Mater. 2024;36:e2401304.
89. Gan L, Lu T, Lu Y, et al. Endosialin-positive CAFs promote hepatocellular carcinoma progression by suppressing CD8+ T cell infiltration. J Immunother Cancer. 2024;12:e009111.
90. Saatci O, Kaymak A, Raza U, et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat Commun. 2020;11:2416.
91. Lee CJ, Jang TY, Jeon SE, et al. The dysadherin/MMP9 axis modifies the extracellular matrix to accelerate colorectal cancer progression. Nat Commun. 2024;15:10422.
92. Sun X, Wu B, Chiang HC, et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature. 2021;599:673-8.
93. Zhang G, Guo L, Yang C, et al. A novel role of breast cancer-derived hyaluronan on inducement of M2-like tumor-associated macrophages formation. Oncoimmunology. 2016;5:e1172154.
94. Blair AB, Wang J, Davelaar J, et al. Dual stromal targeting sensitizes pancreatic adenocarcinoma for anti-programmed cell death protein 1 therapy. Gastroenterology. 2022;163:1267-80.e7.
95. Ishihara J, Fukunaga K, Ishihara A, et al. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci Transl Med. 2017;9:eaan0401.
96. Bhandari C, Moffat A, Shah N, et al. PD-L1 immune checkpoint targeted photoactivable liposomes (iTPALs) prime the stroma of pancreatic tumors and promote self-delivery. Adv Healthc Mater. 2024;13:e2304340.
97. Fattori S, Le Roy A, Houacine J, et al. CD25high effector regulatory T cells hamper responses to PD-1 blockade in triple-negative breast cancer. Cancer Res. 2023;83:3026-44.
98. Redin E, Garmendia I, Lozano T, et al. SRC family kinase (SFK) inhibitor dasatinib improves the antitumor activity of anti-PD-1 in NSCLC models by inhibiting Treg cell conversion and proliferation. J Immunother Cancer. 2021;9:e001496.
99. Meng Y, Ye F, Nie P, et al. Immunosuppressive CD10+ALPL+ neutrophils promote resistance to anti-PD-1 therapy in HCC by mediating irreversible exhaustion of T cells. J Hepatol. 2023;79:1435-49.
100. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369-82.
101. Larroquette M, Guegan JP, Besse B, et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J Immunother Cancer. 2022;10:e003890.
102. Cousin S, Guégan JP, Shitara K, et al. Identification of microenvironment features associated with primary resistance to anti-PD-1/PD-L1 + antiangiogenesis in gastric cancer through spatial transcriptomics and plasma proteomics. Mol Cancer. 2024;23:197.
103. Ajith A, Mamouni K, Horuzsko DD, et al. Targeting TREM1 augments antitumor T cell immunity by inhibiting myeloid-derived suppressor cells and restraining anti-PD-1 resistance. J Clin Invest. 2023;133:e167951.
104. Prasad M, Zorea J, Jagadeeshan S, et al. MEK1/2 inhibition transiently alters the tumor immune microenvironment to enhance immunotherapy efficacy against head and neck cancer. J Immunother Cancer. 2022;10:e003917.
105. Voissière A, Gomez-Roca C, Chabaud S, et al. The CSF-1R inhibitor pexidartinib affects FLT3-dependent DC differentiation and may antagonize durvalumab effect in patients with advanced cancers. Sci Transl Med. 2024;16:eadd1834.
106. Yue T, Li J, Zhu J, et al. Hydrogen sulfide creates a favorable immune microenvironment for colon cancer. Cancer Res. 2023;83:595-612.
107. Boelaars K, Goossens-Kruijssen L, Wang D, et al. Unraveling the impact of sialic acids on the immune landscape and immunotherapy efficacy in pancreatic cancer. J Immunother Cancer. 2023;11:e007805.
108. Bell HN, Huber AK, Singhal R, et al. Microenvironmental ammonia enhances T cell exhaustion in colorectal cancer. Cell Metab. 2023;35:134-49.e6.
109. Zhang J, Wang S, Guo X, et al. Arginine supplementation targeting tumor-killing immune cells reconstructs the tumor microenvironment and enhances the antitumor immune response. ACS Nano. 2022;16:12964-78.
110. Zang J, Yang Y, Zheng X, et al. Dynamic tagging to drive arginine nano-assembly to metabolically potentiate immune checkpoint blockade therapy. Biomaterials. 2023;292:121938.
111. Jin XK, Zhang SM, Liang JL, et al. A PD-L1-targeting regulator for metabolic reprogramming to enhance glutamine inhibition-mediated synergistic antitumor metabolic and immune therapy. Adv Mater. 2024;36:e2309094.
112. Wang W, Zheng H, Jiang J, et al. Engineering micro oxygen factories to slow tumour progression via hyperoxic microenvironments. Nat Commun. 2022;13:4495.
113. Chen N, Li Z, Liu H, et al. Enhancing PD-1 blockade in NSCLC: reprogramming tumor immune microenvironment with albumin-bound statins targeting lipid rafts and mitochondrial respiration. Bioact Mater. 2025;49:140-53.
114. Liu X, Bao X, Hu M, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588:693-8.
115. Li L, Zeng X, Chao Z, et al. Targeting Alpha-ketoglutarate disruption overcomes immunoevasion and improves PD-1 blockade immunotherapy in renal cell carcinoma. Adv Sci. 2023;10:e2301975.
116. Zheng X, Chen J, Deng M, et al. G3BP1 and SLU7 jointly promote immune evasion by downregulating MHC-I via PI3K/Akt activation in bladder cancer. Adv Sci. 2024;11:e2305922.
117. Cheng C, Zha Q, Sun L, et al. VCP downstream metabolite glycerol-3-phosphate (G3P) inhibits CD8+T cells function in the HCC microenvironment. Signal Transduct Target Ther. 2025;10:26.
118. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36:471-82.
119. Giuffrida L, Sek K, Henderson MA, et al. IL-15 preconditioning augments CAR T cell responses to checkpoint blockade for improved treatment of solid tumors. Mol Ther. 2020;28:2379-93.
120. Hegewisch-Becker S, Mendez G, Chao J, et al. First-line nivolumab and relatlimab plus chemotherapy for gastric or gastroesophageal junction adenocarcinoma: the phase II RELATIVITY-060 study. J Clin Oncol. 2024;42:2080-93.
121. Son W, Lee Y, Park Y, et al. Fc-competent TIGITx4-1BB bispecific antibody exerts potent long-lasting antitumor activity by potentiating CD8+ T cell activity and Fcγ receptor-mediated modulation of the tumor microenvironment. J Immunother Cancer. 2025;13:e010728.
122. Hu Y, Paris S, Bertolet G, et al. Combining a nanoparticle-mediated immunoradiotherapy with dual blockade of LAG3 and TIGIT improves the treatment efficacy in anti-PD1 resistant lung cancer. J Nanobiotechnology. 2022;20:417.
123. Dolan M, Libby KA, Ringel AE, van Galen P, McAllister SS. Ageing, immune fitness and cancer. Nat Rev Cancer. 2025;25:848-72.
124. Anobile DP, Salaroglio IC, Tabbò F, et al. Autocrine 17-β-estradiol/estrogen receptor-α loop determines the response to immune checkpoint inhibitors in non-small cell lung cancer. Clin Cancer Res. 2023;29:3958-73.
125. Chakraborty B, Byemerwa J, Shepherd J, et al. Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J Clin Invest. 2021;131:e151347.
126. Adurthi S, Kumar MM, Vinodkumar HS, et al. Oestrogen receptor-α binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci Rep. 2017;7:17289.
127. Svoronos N, Perales-Puchalt A, Allegrezza MJ, et al. Tumor cell-independent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov. 2017;7:72-85.
128. Milette S, Hashimoto M, Perrino S, et al. Sexual dimorphism and the role of estrogen in the immune microenvironment of liver metastases. Nat Commun. 2019;10:5745.
129. Artham S, Juras PK, Goyal A, et al. Estrogen signaling suppresses tumor-associated tissue eosinophilia to promote breast tumor growth. Sci Adv. 2024;10:eadp2442.
130. Kwon Y. Potential pro-tumorigenic effect of bisphenol A in breast cancer via altering the tumor microenvironment. Cancers. 2022;14:3021.
132. Yuan B, Clark CA, Wu B, et al. Estrogen receptor beta signaling in CD8+ T cells boosts T cell receptor activation and antitumor immunity through a phosphotyrosine switch. J Immunother Cancer. 2021;9:e001932.
133. Guan X, Polesso F, Wang C, et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature. 2022;606:791-6.
134. Kwon H, Schafer JM, Song NJ, et al. Androgen conspires with the CD8+ T cell exhaustion program and contributes to sex bias in cancer. Sci Immunol. 2022;7:eabq2630.
135. Liu Q, You B, Meng J, et al. Targeting the androgen receptor to enhance NK cell killing efficacy in bladder cancer by modulating ADAR2/circ_0001005/PD-L1 signaling. Cancer Gene Ther. 2022;29:1988-2000.
136. Jin WB, Xiao L, Jeong M, et al.; JRI IBD Live Cell Bank Consortium. Microbiota-derived bile acids antagonize the host androgen receptor and drive anti-tumor immunity. Cell. 2025;188:2336-53.e38.
137. Madi A, Shi H, Su M, et al. DGAT1 mediates sex-specific CD8+ T cell antitumour responses. Nat Metab. 2026;8:685-703.
138. Zitvogel L, Fidelle M, Kroemer G. Long-distance microbial mechanisms impacting cancer immunosurveillance. Immunity. 2024;57:2013-29.
139. Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084-9.
140. Fidelle M, Rauber C, Alves Costa Silva C, et al. A microbiota-modulated checkpoint directs immunosuppressive intestinal T cells into cancers. Science. 2023;380:eabo2296.
141. Park JS, Gazzaniga FS, Wu M, et al. Targeting PD-L2-RGMb overcomes microbiome-related immunotherapy resistance. Nature. 2023;617:377-85.
142. Najar TA, Hao Y, Hao Y, et al. Microbiota-induced T cell plasticity enables immune-mediated tumour control. Nature. 2026;651:201-10.
143. Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91-7.
144. Davar D, Dzutsev AK, McCulloch JA, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371:595-602.
145. Zhou H, Sun R, Nie X, et al. A clinic-responder-derived defined microbial consortium enhances anti-PD-1 immunotherapy efficacy in mice. Nat Microbiol. 2026;11:993-1007.
146. Fu A, Yao B, Dong T, et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell. 2022;185:1356-72.e26.
147. Sahler JM, Eade CR, Altier C, March JC. Salmonella enterica serovar typhimurium increases functional PD-L1 synergistically with gamma interferon in intestinal epithelial cells via Salmonella pathogenicity island 2. Infect Immun. 2018;86:e00674-17.
148. Wu YY, Lin CW, Cheng KS, et al. Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin Exp Immunol. 2010;161:551-9.
149. Hezaveh K, Shinde RS, Klötgen A, et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity. 2022;55:324-40.e8.
150. Nejman D, Livyatan I, Fuks G, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020;368:973-80.
151. Chen J, Gao Y, Chen Y, et al. Identification and validation of intratumoral microbiome associated with sensitization to immune checkpoint inhibitors. Cell Rep Med. 2025;6:102306.
152. Canale FP, Basso C, Antonini G, et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature. 2021;598:662-6.
153. Murphy S, Rahmy S, Gan D, et al. Ketogenic diet alters the epigenetic and immune landscape of prostate cancer to overcome resistance to immune checkpoint blockade therapy. Cancer Res. 2024;84:1597-612.
154. Fiuza-Luces C, Valenzuela PL, Gálvez BG, et al. The effect of physical exercise on anticancer immunity. Nat Rev Immunol. 2024;24:282-93.
155. Geng Q, Li L, Shen Z, et al. Norepinephrine inhibits CD8+ T-cell infiltration and function, inducing anti-PD-1 mAb resistance in lung adenocarcinoma. Br J Cancer. 2023;128:1223-35.
156. Qiao G, Chen M, Mohammadpour H, et al. Chronic adrenergic stress contributes to metabolic dysfunction and an exhausted phenotype in T cells in the tumor microenvironment. Cancer Immunol Res. 2021;9:651-64.
157. Yan X, Liu P, Li D, et al. Novel evidence for the prognostic impact of β-blockers in solid cancer patients receiving immune checkpoint inhibitors. Int Immunopharmacol. 2022;113:109383.
158. Zhu J, Naulaerts S, Boudhan L, Martin M, Gatto L, Van den Eynde BJ. Tumour immune rejection triggered by activation of α2-adrenergic receptors. Nature. 2023;618:607-15.
159. Yang J, Wu Y, Lv X, et al. Neurotransmitters: an emerging target for therapeutic resistance to tumor immune checkpoint inhibitors. Mol Cancer. 2025;24:216.
160. Schneider MA, Heeb L, Beffinger MM, et al. Attenuation of peripheral serotonin inhibits tumor growth and enhances immune checkpoint blockade therapy in murine tumor models. Sci Transl Med. 2021;13:eabc8188.
161. Balood M, Ahmadi M, Eichwald T, et al. Nociceptor neurons affect cancer immunosurveillance. Nature. 2022;611:405-12.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.