


Impact of autoimmune phenomena on the clinical presentation and prognosis of Wilson’s disease: a retrospective cohort study
 0
0 Abstract
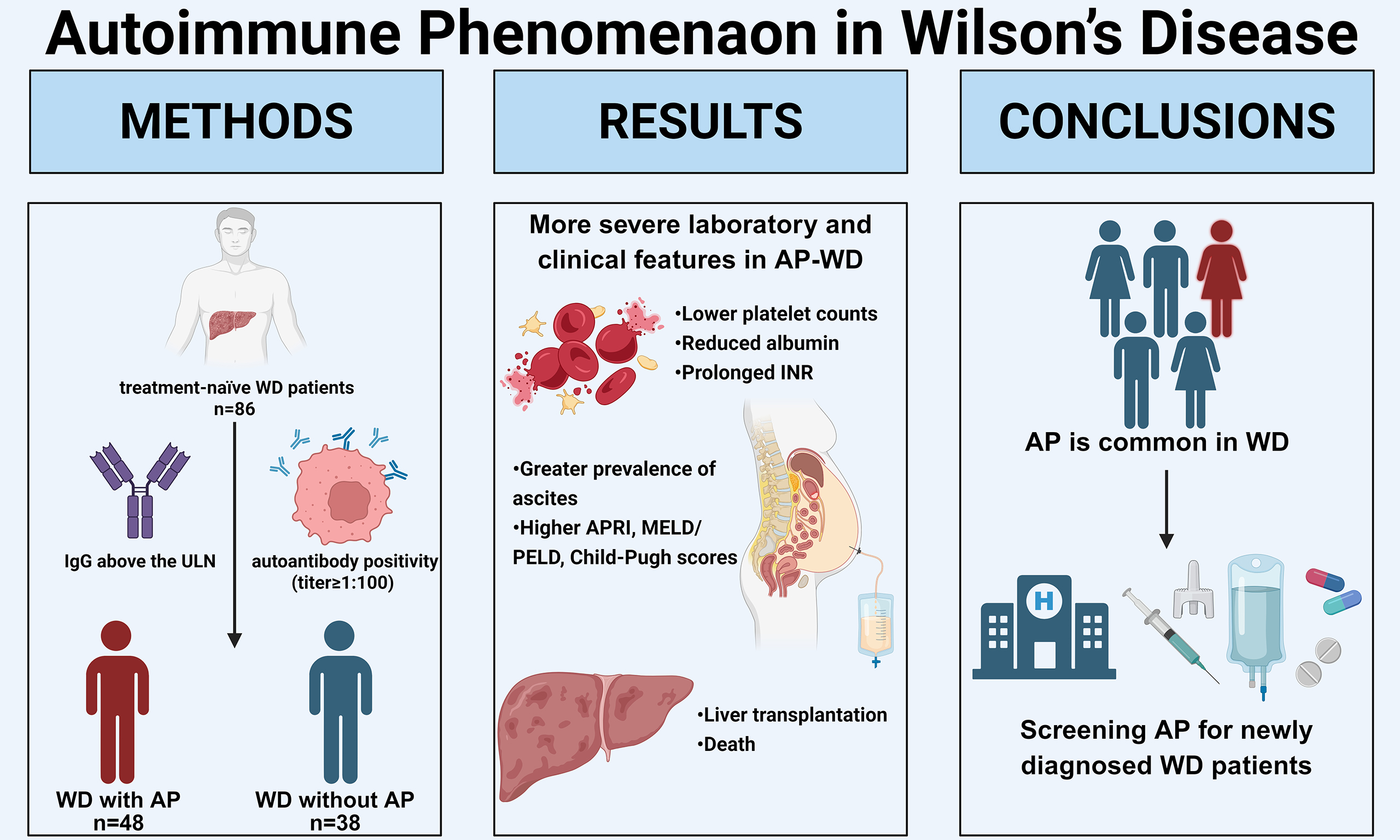
Aim: To investigate the clinical relevance of autoimmune phenomena (AP), including autoantibody positivity and elevated serum immunoglobulins, in patients with Wilson’s disease (WD), particularly with respect to disease severity and prognosis.
Methods: We retrospectively enrolled treatment-naïve WD patients (Leipzig score ≥ 4) and classified them as WD with AP (AP-WD) or WD without AP (NAP-WD) based on autoantibody positivity (titer ≥ 1:100) and/or immunoglobulin G (IgG) above the upper limit of normal. Baseline laboratory data, clinical complications, and liver histopathology were compared. Patients received standard anti-copper therapy and were followed longitudinally to evaluate the impact of AP on liver-related outcomes.
Results: Eighty-six treatment-naïve WD patients were included (48 AP-WD, 38 NAP-WD). At baseline, AP-WD patients exhibited more severe laboratory and clinical features, including lower platelet counts, reduced albumin, prolonged international normalized ratio, higher aspartate aminotransferase-to-platelet ratio index (APRI), Model for End-Stage Liver Disease (MELD)/Pediatric End-Stage Liver Disease (PELD), and Child-Pugh scores, and greater prevalence of ascites (all P < 0.05). Histopathological analysis demonstrated increased plasma cell infiltration and heightened portal inflammatory activity in the AP-WD group (P < 0.05). Longitudinal follow-up revealed that the presence of AP was independently associated with an increased risk of adverse liver-related events, including liver transplantation, or death.
Conclusion: AP is common in WD and correlates with more severe hepatic dysfunction and poorer long-term outcomes. Screening for autoantibodies and IgG levels in newly diagnosed WD patients may provide important prognostic insight and facilitate early risk stratification, guiding tailored monitoring and management strategies.
Keywords
INTRODUCTION
Wilson’s disease (WD), also known as hepatolenticular degeneration, is a rare autosomal recessive disorder caused by mutations in the ATPase copper transporting beta (ATP7B) gene located on chromosome 13q14. The ATP7B gene encodes a copper-transporting P-type ATPase that plays a crucial role in maintaining copper homeostasis by mediating its incorporation into ceruloplasmin and promoting biliary copper excretion[1]. Pathogenic variants in ATP7B disrupt these functions and lead to systemic copper accumulation, primarily affecting the liver, brain, and kidneys. The progressive retention of copper results in multisystem damage, with clinical manifestations mainly characterized by hepatic and neurological dysfunction[2]. Early diagnosis and timely initiation of anti-copper therapy are essential to improve long-term outcomes[3-5].
In recent years, increasing attention has been given to the occurrence of autoimmune phenomena (AP) in patients with WD. According to the definition proposed in the drug-induced liver injury (DILI) guideline[6], AP refer to immune-related abnormalities such as the presence of circulating autoantibodies or autoimmune hepatitis (AIH)-like features that coexist with WD but are not necessarily indicative of a primary autoimmune liver disease. Several studies have reported a higher prevalence of positive autoantibodies, including antinuclear antibodies (ANA), smooth muscle antibodies (SMA), and anti-neutrophil cytoplasmic antibodies (ANCA), in patients with WD compared with the general population[7,8]. Some patients even present with AIH-like characteristics, such as elevated serum immunoglobulin G (IgG) levels and interface hepatitis on liver biopsy[9-11]. However, the reported frequency and clinical significance of these autoimmune markers vary considerably among studies.
Previous research suggested that the presence of autoantibodies may not correlate with liver function, copper metabolism indicators, or liver stiffness in WD patients[8], but this finding was limited by small sample sizes and a lack of long-term follow-up. Furthermore, comprehensive data on the epidemiological characteristics, clinical correlations, and prognostic implications of AP in WD remain scarce, particularly among Chinese patients, whose genetic and environmental backgrounds may differ from those of Western populations.
Given these uncertainties, we conducted a retrospective cohort study to investigate the prevalence of AP and their association with clinical features, disease severity, and prognosis in patients with WD. We aimed to quantify autoantibody positivity rates, characterize antibody profiles, and measure IgG levels in patients with WD, and to evaluate their associations with clinical features, disease prognosis, and histopathological findings, in order to determine whether the presence of autoimmune markers defines a distinct clinical phenotype with prognostic significance.
METHODS
Patients population
This single-center retrospective cohort study included patients with WD hospitalized at Beijing YouAn Hospital, Capital Medical University, from August 2008 to July 2024. Diagnosis was based on the Leipzig scoring system (score ≥ 4)[12]. ATP7B gene mutations were detected using next-generation sequencing. Kayser-Fleischer (KF) rings were independently assessed by two experienced ophthalmologists, and any discrepancies were resolved by consensus with a third ophthalmologist. All enrolled patients had complete clinical data at the time of enrollment, including blood counts, biochemistry, coagulation profiles, abdominal ultrasound, and computed tomography (CT) imaging.
Exclusion criteria were as follows: (1) Presence of other liver diseases, including but not limited to hepatitis B or C virus infection, DILI, alcoholic liver disease, or hepatocellular carcinoma; (2) Prior treatment with anti-copper agents (e.g., penicillamine, trientine, zinc salts); (3) Absence of baseline autoantibody or immunoglobulin measurements.
Clinical assessment, definitions, and follow-up
Clinical data were collected at baseline, including demographics (age, sex), presence of KF rings, routine laboratory parameters (complete blood count, liver function tests, coagulation profile, and biochemical markers), copper metabolism indices (serum ceruloplasmin, 24 h urinary copper excretion), autoantibody profiles [ANA, anti-smooth muscle antibody (ASMA), anti-mitochondrial antibody (AMA), and anti-cytoskeletal antibody (CS)], immunoglobulin levels [IgG, immunoglobulin A (IgA), and immunoglobulin M (IgM)], and imaging findings (abdominal ultrasound and CT). The aspartate aminotransferase-to-platelet ratio index (APRI), Child-Pugh score, and Model for End-Stage Liver Disease (MELD) or Pediatric End-Stage Liver Disease (PELD) scores (using MELD for patients aged ≥ 12 years and PELD for those aged < 12 years) were calculated manually according to standard formulas.
Cirrhosis was diagnosed based on the 2018 European Association for the Study of the Liver (EASL) Clinical Practice Guideline[13], incorporating clinical, biochemical, imaging, and, when available, histological criteria. Liver failure was defined to include acute liver failure (ALF), acute-on-chronic liver failure (ACLF), and chronic liver failure (CLF), allowing comprehensive coverage of all clinically relevant liver failure phenotypes in WD. The diagnosis of ALF, ACLF, and CLF was made according to the 2024 Chinese Guidelines for the Diagnosis and Treatment of Liver Failure[14].
Patients with positive autoantibodies (titer ≥ 1:100)[15] and/or serum IgG levels above the upper limit of normal (ULN; 17.4 g/L) were defined as having WD with AP (AP-WD), whereas those with negative autoantibodies and normal IgG levels were classified as WD without AP (NAP-WD). Liver-related adverse events (LRAEs) were defined as occurrence of liver transplantation or death, and were designated as the primary endpoint.
All patients received standard anti-copper therapy according to the clinical practice guideline for the management of WD[16] immediately upon diagnostic confirmation. Follow-up data were obtained through a combination of outpatient visits and structured telephone interviews to capture longitudinal clinical outcomes. For time-to-event analyses, follow-up was administratively censored at a maximum of 60 months.
Histological assessment
To assess the impact of AP on hepatic pathology, liver tissue specimens were obtained from 17 patients in the study cohort through percutaneous biopsy. All samples underwent routine staining with hematoxylin-eosin (HE), Masson’s trichrome, reticulin, cytokeratin 19 (CK19), and special stains for iron and copper.
All slides were independently reviewed by an experienced hepatic pathologist blinded to patients’ immunological data. The evaluation focused on histological features suggestive of AIH, including interface hepatitis, plasma cell infiltration, and hepatocellular rosette formation. In addition, general histological characteristics were assessed, including the degree of steatosis, hepatocyte ballooning, Mallory-Denk bodies, portal and lobular inflammation, and fibrosis stage. Portal interface hepatitis and inflammation within the portal areas were quantitatively assessed using the Ishak scoring system[17]. Plasma cell infiltration in lobular or portal regions was graded on a 0-3 scale: 0 (absent), 1 [1-4 cells per high-power field (HPF)], 2 (5-9 cells/HPF), and 3 (≥ 10 cells/HPF)[18]. In addition, the Non-Alcoholic Fatty Liver Disease (NAFLD) Activity Score (NAS) was calculated for each patient, comprising steatosis (0-3), lobular inflammation (0-3), and hepatocellular ballooning (0-2)[19]. Fibrosis was staged via the Scheuer system: S0: No fibrosis; S1: Portal fibrosis; S2: Perportal fibrosis or portal-portal (P-P) fibrous septa (intact lobular architecture); S3: Bridging/septal fibrosis (structural damage without overt cirrhosis); S4: Cirrhosis (pseudolobule formation).
Statistical analysis
Statistical analyses and figure preparation were performed using SPSS version 26.0 (IBM Corp., Armonk, NY, USA), R version 4.3.0 (R Foundation for Statistical Computing, Vienna, Austria), and GraphPad Prism version 10 (GraphPad Software, San Diego, CA, USA). Continuous variables with a normal distribution were expressed as mean ± standard deviation (SD), with comparisons between two groups conducted using Student’s t-test and comparisons among multiple groups performed using analysis of variance (ANOVA). Non-normally distributed continuous variables were presented as median [interquartile range (IQR), Q25-Q75] and compared using the rank-sum test for two or more groups. Categorical variables were expressed as counts and percentages. Due to the retrospective nature of the cohort, some patients had missing data; therefore, all percentages and positivity rates were calculated based on the number of patients who underwent the respective tests rather than the total cohort. Group differences were assessed using the χ2 test. Spearman’s rank correlation was used to assess associations between continuous variables. The cumulative risk was estimated using the Kaplan-Meier method, and the log-rank test was employed to evaluate the differences between the groups. In addition, a univariable Cox proportional hazards model was used to estimate hazard ratios (HRs) with corresponding 95% confidence intervals (CI). A two-sided P value < 0.05 was considered statistically significant.
RESULTS
Demographics and clinical characteristics at baseline
A total of 204 hospitalized patients with WD were initially screened. After applying exclusion criteria, removing 6 patients with concurrent liver disorders, 98 with prior anti-copper therapy, and 14 without autoantibody or immunoglobulin measurements. A total of 86 treatment-naïve patients were included in the final analysis [Figure 1].
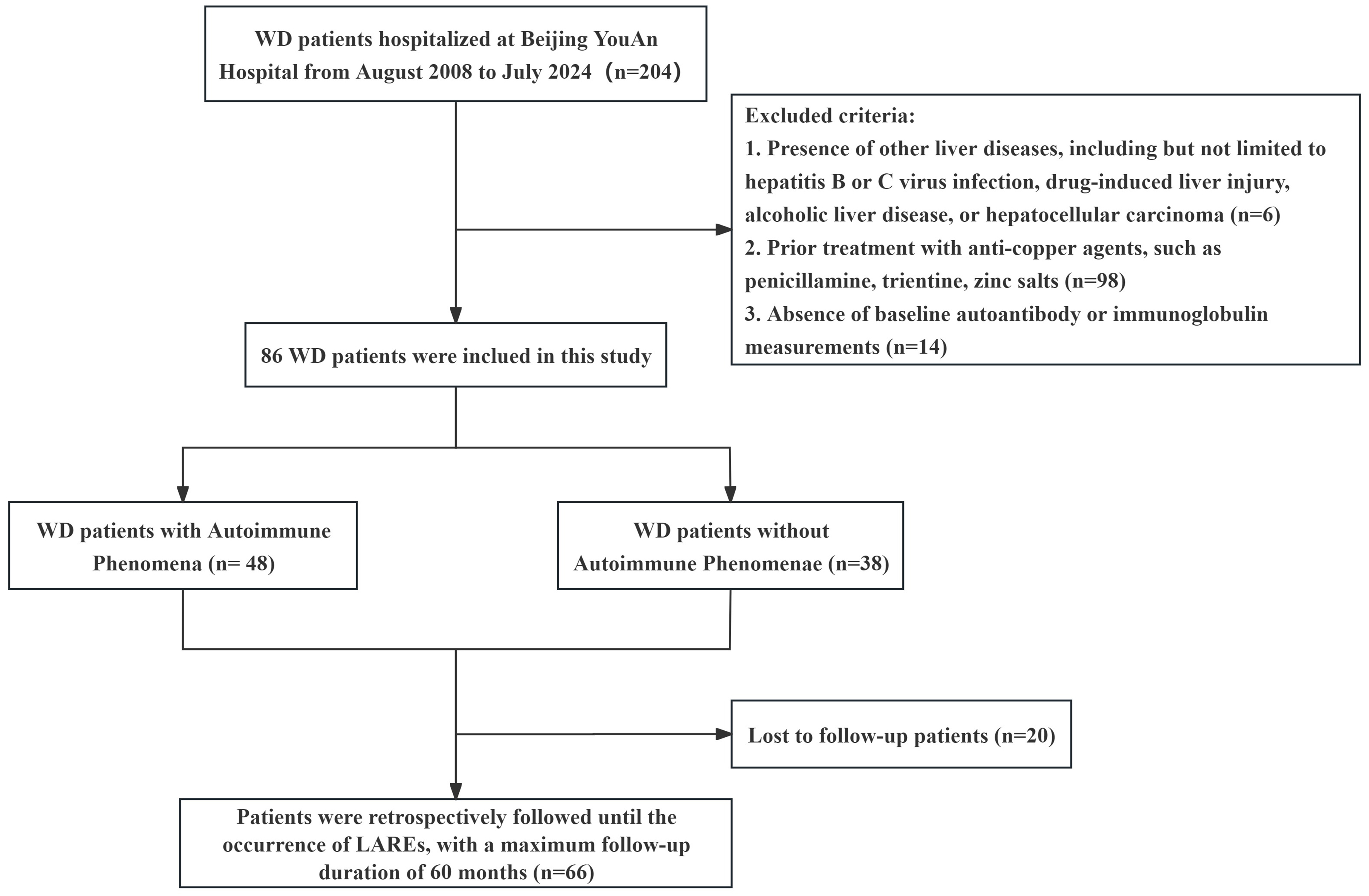
Figure 1. Flowchart illustrating patients selection and classification. Generated using WPS Office. WD: Wilson’s disease; LRAEs: liver-related adverse events.
At the time of initial diagnosis, the median age of patients was 18 years (11.0-33.8), and 42 patients (48.8%) were male. Most patients exhibited mild elevations in liver enzymes, with abnormal ceruloplasmin levels and 24 h urinary copper excretion at diagnosis. KF rings were observed in 51 patients (59.3%). Based on predefined autoimmune criteria, 48 patients (55.8%) were classified as AP-WD. Among these, 25 patients had isolated autoantibody positivity, 7 had isolated IgG elevation (> 17.4 g/L), and 16 exhibited both autoantibody positivity and elevated IgG levels. The majority (n = 53, 61.6%) showed evidence of cirrhosis on imaging or histopathology. Detailed patient characteristics are summarized in Table 1.
Characteristics of patients with WD at baseline
| Variables | n = 86 |
| Age (years) | 18.00 (11.00, 33.75) |
| Sex, males, n (%) | 42 (48.84) |
| Presence of K-F rings, n (%) | 51 (59.30) |
| Liver cirrhosis, n (%) | 53 (61.63) |
| Compensated cirrhosis | 18 (20.93) |
| Decompensated cirrhosis | 35 (40.70) |
| Complication, n (%) | |
| Splenomegaly | 57 (66.28) |
| Ascites | 35 (40.70) |
| Gastroesophageal varices | 19 (22.09) |
| Laboratory tests | |
| WBC (109/L) | 5.28 (4.00, 6.50) |
| NEUT (109/L) | 2.67 (1.85, 3.58) |
| RBC (1012/L) | 3.87 (2.91, 4.63) |
| Hb (g/L) | 119.00 (94.00, 130.75) |
| PLT (109/L) | 123.50 (73.25, 209.00) |
| PT (S) | 14.15 (11.60, 20.18) |
| INR | 1.26 (1.03, 1.88) |
| PTA (%) | 66.55 (36.45, 91.25) |
| APTT (S) | 39.75 (33.55, 49.75) |
| FIB (g/L) | 1.75 ± 0.60 |
| ALT (U/L) | 51.80 (23.73, 98.72) |
| AST (U/L) | 62.05 (38.70, 106.68) |
| γ-GT (U/L) | 67.00 (42.90, 111.08) |
| TBil (μmol/L) | 25.35 (13.45, 64.02) |
| DBil (μmol/L) | 9.60 (4.12, 33.52) |
| TP (g/L) | 62.85 ± 8.29 |
| ALB (g/L) | 34.58 ± 7.90 |
| AFP (ng/mL) | 11.05 (3.86, 86.39) |
| Cr (μmol/L) | 49.80 (37.25, 64.50) |
| UREA (mmol/L) | 4.33 (3.54, 5.53) |
| CHOL (mmol/L) | 3.53 ± 1.28 |
| ALP (U/L) | 116.35 (78.25, 230.90) |
| CHE (U/L) | 3,046.50 (2,146.30, 4,988.00) |
| IgG (g/L) | 14.58 ± 4.95 |
| IgA (g/L) | 2.89 (1.96, 3.59) |
| IgM (g/L) | 1.52 (1.01, 2.09) |
| Ceruloplasmin (g/L) | 0.05 (0.03, 0.09) |
| Urinary copper (μg/24h) | 380.79 (143.06, 1,294.84) |
| Autoantibodies positive, n (%) | 41 (50.62) |
| IgG elevation(> 17.4 g/L), n (%) | 17 (20.24) |
| Scores | |
| APRI | 1.31 (0.79, 2.25) |
| Child-Pugh | 6.00 (5.00, 11.00) |
| MELD | 10.50 (8.00, 15.00) |
| PELD | -15.00 (-29.25, 15.00) |
Autoantibody expression patterns and titers in WD patients
We further assessed the prevalence and patterns of autoantibodies in WD patients. Of the 86 patients included in the study, 81 underwent autoantibody testing, and 41 (50.6%) were found to be autoantibody-positive. Notably, this positivity rate was substantially higher than that reported in the general population (10%-20%)[20]. Among the 41 autoantibody-positive WD patients, 40 (97.6%) were positive for ANA, corresponding to 49.4% of the 81 tested patients. Of these, 37 (45.7% of the 81 tested patients) had isolated ANA positivity. One patient (1.2%) was positive for both ANA and ASMA, one (1.2%) for ANA and AMA, one (1.2%) for ANA and CS, and one (1.2%) for ASMA and CS. As summarized in Table 2, most WD patients with AP exhibited low-titer autoantibodies.
Immunofluorescent antibody patterns and titers found in patients with WD
| Pattern | Type | Titer | Total | ||
| 1:100 | 1:320 | 1:1,000 | |||
| ANA | - | 30 | 6 | 4 | 40 |
| Speckled | Nuclear | 15 | 1 | 1 | 17 |
| Homogeneous | Nuclear | 0 | 0 | 0 | 0 |
| Nucleolar | Nuclear | 1 | 1 | 0 | 2 |
| Speckled | Cytoplasmic | 0 | 0 | 0 | 0 |
| Golgi | Cytoplasmic | 1 | 0 | 0 | 1 |
| Mixed | - | 3 | 2 | 1 | 6 |
| Unknown | - | 10 | 2 | 2 | 14 |
| ASMA | - | 1 | 1 | 0 | 2 |
| CS | - | 1 | 1 | 0 | 2 |
| AMA | - | 0 | 1 | 0 | 1 |
Comparison of baseline clinical features between WD patients with and without AP
To compare baseline clinical characteristics between WD patients with and without AP, we systematically compared demographic parameters, laboratory indicators, liver-related scores, and clinical complications between AP-WD and NAP-WD patients [Table 3]. At diagnosis, there were no significant differences in age or sex between the two groups, confirming their baseline comparability.
Baseline characteristics of treatment-naïve WD patients with and without AP
| Variables | NAP-WD group (n = 38) | AP-WD group (n = 48) | Statistic | P |
| Age (years) | 16.50 (11.00, 27.50) | 25.00 (10.50, 39.25) | Z = -1.62 | 0.106 |
| Sex, males, n (%) | 23 (60.53) | 19 (39.58) | χ2 = 3.72 | 0.054 |
| Presence of K-F rings, n (%) | 16 (42.11) | 35 (72.92) | χ2 = 9.02 | 0.011 |
| Liver cirrhosis, n (%) | 17 (44.74) | 36 (75.00) | χ2 = 8.21 | 0.004 |
| Complication, n (%) | ||||
| Splenomegaly | 21 (55.26) | 36 (75.00) | - | 0.230 |
| Ascites | 9 (23.68) | 26 (54.17) | - | 0.013 |
| Gastroesophageal varices | 5 (13.16) | 14 (29.17) | χ2 = 3.16 | 0.076 |
| Laboratory tests | ||||
| WBC (109/L) | 5.14 (4.25, 6.50) | 5.29 (3.68, 6.63) | Z = -0.30 | 0.767 |
| NEUT (109/L) | 2.65 (2.08, 3.40) | 2.72 (1.81, 3.71) | Z = -0.37 | 0.715 |
| RBC (1012/L) | 4.52 (3.75, 4.94) | 3.51 (2.75, 4.17) | Z = -3.49 | < 0.001 |
| Hb (g/L) | 125.00 (106.25, 140.75) | 108.50 (82.00, 127.25) | Z = -3.03 | 0.002 |
| PLT (109/L) | 157.00 (121.50, 265.75) | 94.00 (63.50, 146.75) | Z = -3.39 | < 0.001 |
| PT (S) | 11.90 (11.03, 14.45) | 16.85 (13.57, 23.82) | Z = -3.30 | < 0.001 |
| INR | 1.06 (0.95, 1.27) | 1.56 (1.23, 2.08) | Z = -4.39 | < 0.001 |
| PTA (%) | 88.15 (68.50, 103.63) | 49.50 (32.62, 68.25) | Z = -4.57 | < 0.001 |
| APTT (S) | 34.35 (31.90, 41.50) | 42.75 (37.55, 53.70) | Z = -3.57 | < 0.001 |
| FIB (g/L) | 2.05 (1.68, 2.50) | 1.56 (1.18, 1.93) | Z = -3.57 | < 0.001 |
| AFP (ng/mL) | 10.26 (3.03, 334.49) | 11.60 (4.59, 38.55) | Z = -0.24 | 0.811 |
| ALT (U/L) | 70.70 (30.23, 104.25) | 45.65 (19.05, 69.75) | Z = -2.31 | 0.021 |
| AST (U/L) | 54.90 (39.03, 97.75) | 75.40 (38.40, 107.93) | Z = -1.00 | 0.319 |
| γ-GT (U/L) | 67.35 (37.85, 97.62) | 66.50 (46.50, 113.22) | Z = -0.49 | 0.623 |
| TBil (μmol/L) | 23.00 (10.67, 36.70) | 36.20 (19.50, 97.10) | Z = -2.20 | 0.027 |
| DBil (μmol/L) | 6.90 (2.42, 13.90) | 14.90 (5.60, 51.05) | Z = -2.61 | 0.009 |
| ALP (U/L) | 128.50 (91.70, 248.72) | 115.42 (74.75, 170.50) | Z = -1.50 | 0.132 |
| CHE (U/L) | 4,474.00 (2,644.10, 6,169.00) | 2,389.00 (1,588.00, 3,507.00) | Z = -3.81 | < 0.001 |
| TP (g/L) | 64.70 (59.95, 69.27) | 63.90 (55.10, 69.00) | Z = -0.53 | 0.599 |
| ALB (g/L) | 38.69 ± 6.78 | 31.33 ± 7.22 | t = 4.82 | < 0.001 |
| Cr (μmol/L) | 52.45 (37.02, 65.82) | 49.30 (39.42, 62.90) | Z = -0.25 | 0.804 |
| UREA (mmol/L) | 4.35 (3.41, 5.11) | 4.31 (3.57, 5.64) | Z = -0.79 | 0.429 |
| CHOL (mmol/L) | 4.01 ± 1.22 | 3.14 ± 1.21 | t = 3.26 | 0.002 |
| IgG (g/L) | 11.30 (10.17, 13.78) | 17.10 (12.97, 19.68) | Z = -5.01 | < 0.001 |
| IgA (g/L) | 2.52 (1.69, 3.17) | 3.18 (2.14, 3.95) | Z = -2.10 | 0.035 |
| IgM (g/L) | 1.29 (0.94, 1.87) | 1.67 (1.20, 2.45) | Z = -1.81 | 0.070 |
| Ceruloplasmin (g/L) | 0.04 (0.03, 0.08) | 0.06 (0.04, 0.10) | Z = -1.18 | 0.237 |
| Urinary copper (μg/24 h) | 235.20 (92.62, 617.71) | 1,005.07 (239.76, 1,918.01) | Z = -2.36 | 0.018 |
| Scores | ||||
| APRI | 0.93 (0.58, 1.59) | 1.56 (1.01, 3.00) | Z = -3.06 | 0.002 |
| Child-Pugh | 5.00 (5.00, 6.00) | 9.00 (6.00, 11.00) | Z = -3.78 | < 0.001 |
| MELD | 9.00 (6.70, 11.50) | 13.00 (9.00, 18.00) | Z = -2.88 | 0.004 |
| PELD | -28.00 (-31.00, -18.00) | 1.00 (-13.00, 22.00) | Z = -2.23 | 0.026 |
Analysis of routine laboratory parameters revealed that patients with AP-WD exhibited significantly lower red blood cell counts, hemoglobin levels, and platelet counts (P < 0.001, P = 0.002, and P < 0.001, respectively). Coagulation assessments showed prolonged prothrombin time, activated partial thromboplastin time (APTT), and elevated international normalized ratio (INR, all P < 0.001), along with reduced prothrombin time activity (PTA) and fibrinogen levels (both P < 0.001). Liver function tests indicated higher total and direct bilirubin levels (P = 0.027 and P = 0.009, respectively) and lower serum cholinesterase and albumin levels (both P < 0.001) in the AP-WD group. Notably, ALT was slightly higher in the NAP-WD group (P = 0.021).
Regarding copper metabolism, 24 h urinary copper excretion was significantly elevated in AP-WD patients compared to NAP-WD patients (P = 0.018), suggesting greater copper burden in patients with AP. Consistently, liver-related scoring systems reflected greater disease severity in the AP-WD group, with higher APRI, Child-Pugh, and MELD/PELD scores (all P < 0.05). In addition, cirrhosis prevalence and KF ring positivity were significantly higher in AP-WD patients (P = 0.004 and P = 0.011, respectively).
Evaluation of clinical complications showed that ascites occurred more frequently in the AP-WD group than in the NAP-WD group (54.2% vs. 23.7%, P = 0.013), whereas the incidence of splenomegaly or gastroesophageal varices did not differ significantly between the groups (both P > 0.05).
To further explore whether the titer of autoantibodies affected disease severity, subgroup analyses were performed. These analyses revealed no significant differences in age, sex, platelet count, serum albumin, total bilirubin (TBil), APTT, PTA, copper metabolism indicators, liver-related scores, or clinical complications across patients with different autoantibody titers [Supplementary Tables 1 and 2], indicating that the presence of AP, rather than titer levels, may be the key factor associated with more severe baseline liver involvement in WD.
Correlation between serum IgG levels and clinical parameters in WD patients
To further elucidate the relationship between immune activation and hepatic functional impairment, we explored correlations between serum IgG levels and key clinical parameters in WD patients [Figure 2]. Spearman’s correlation analysis revealed that serum albumin and cholinesterase were inversely correlated with IgG levels (both P < 0.05). Similarly, the INR, an indicator of coagulation dysfunction, showed a positive correlation with IgG (P < 0.05). In contrast, no significant correlations were observed between IgG and hemoglobin or MELD scores. These findings suggest that elevated IgG levels in WD are closely linked to hepatic synthetic dysfunction and coagulation abnormalities.
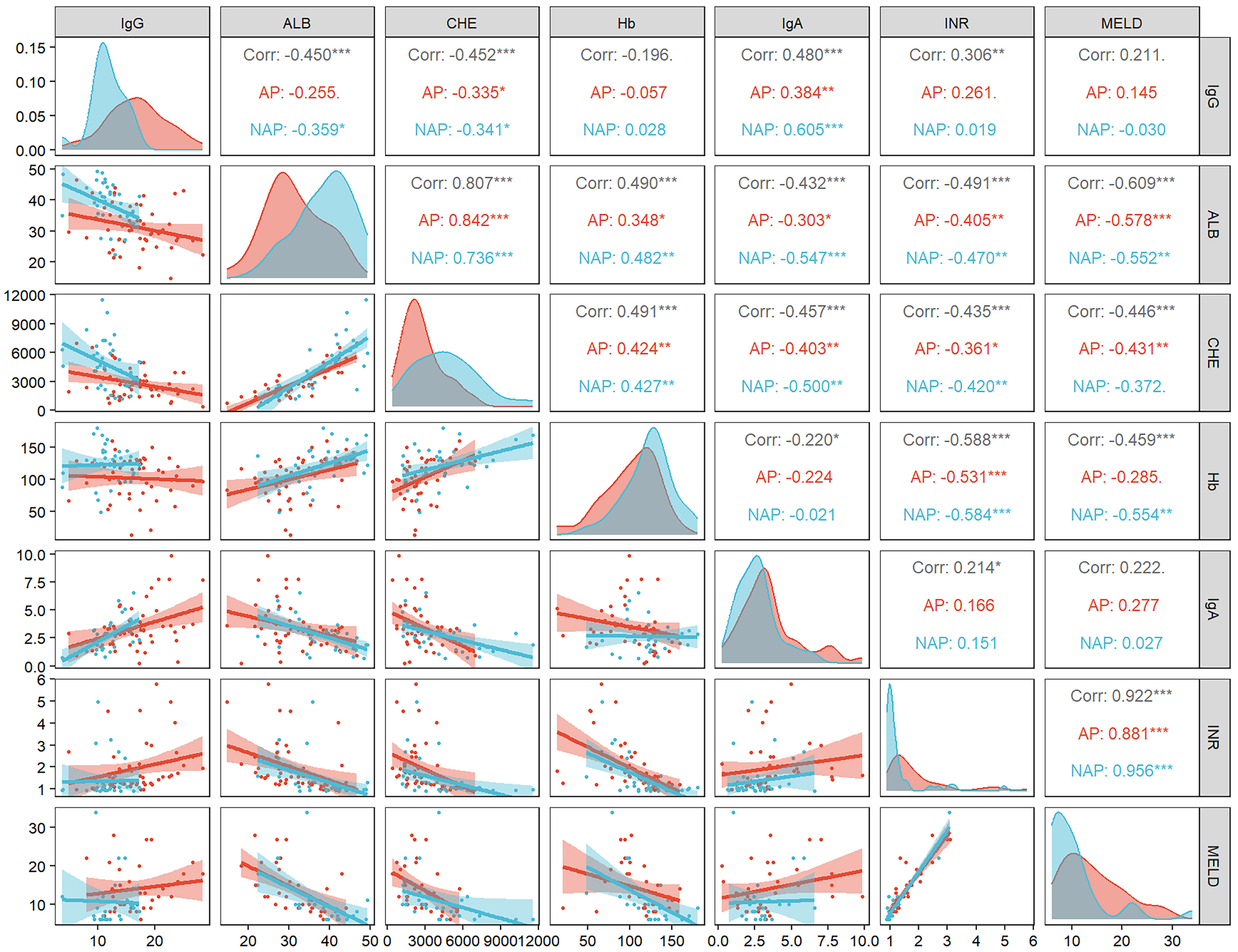
Figure 2. Correlation between IgG levels and liver function-related indicators. The number in the box is Spearman’s correlation coefficient (ρ). *P < 0.05; **P < 0.01; ***P < 0.001. Generated using R 4.3.0. AP: AP-WD group; NAP: NAP-WD group; IgG: Immunoglobulin G; ALB: albumin; CHE: cholinesterase; Hb: hemoglobin; IgA: immunoglobulin A; INR: international normalized ratio; MELD: model for end-stage liver disease; WD: Wilson’s disease.
Comparative histopathological analysis of liver tissue in AP-WD and NAP-WD patients
To further elucidate the histopathological correlates of these biochemical findings, we next compared liver tissue features between AP-WD and NAP-WD patients. A total of 17 percutaneous liver biopsy specimens were included in this study. Microscopic examination revealed that AP-WD patients exhibited more pronounced interface hepatitis and plasma cell infiltration [Figure 3]. Quantitative comparison of histopathological parameters between the AP-WD (n = 6) and NAP-WD (n = 11) groups [Table 4] showed significantly greater plasma cell infiltration (P < 0.05) and higher inflammatory activity in portal areas (P < 0.05) in the AP-WD group. Rosette-like hepatocellular arrangements were more frequent in AP-WD patients (3/6, 50%) than in NAP-WD patients (1/11, 9.1%), though the difference did not reach statistical significance (P = 0.099). No significant differences were observed in other parameters, including NAFLD activity score, fibrosis stage, interface hepatitis, or presence of Mallory-Denk bodies (all P > 0.05). Overall, histopathological evaluation demonstrated more intense portal inflammation and plasma cell infiltration in AP-WD patients, highlighting a distinct immunopathological pattern in treatment-naïve WD with autoimmune features.
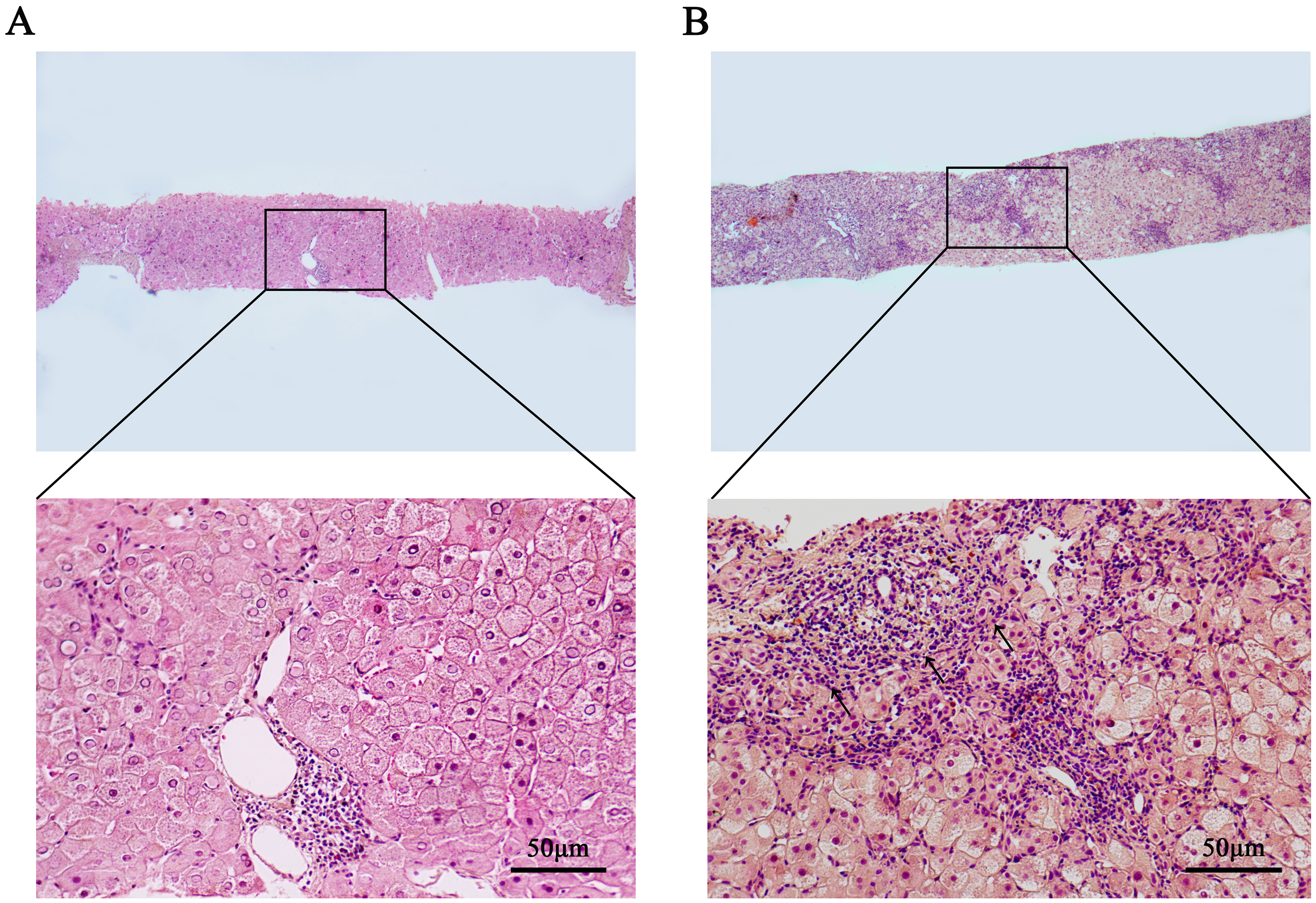
Figure 3. Histopathological features in liver biopsies of NAP-WD and AP-WD patients. (A) NAP-WD patient: no obvious interface hepatitis or plasma cell infiltration was observed (HE staining, 40×/200×); (B) AP-WD patients: marked interface hepatitis and plasma cell infiltration were observed (HE staining, 40×/200×). Diffuse hepatocyte enlargement, with moderate to severe inflammatory cell infiltration within the portal tracts, extensive fibrous tissue proliferation. Disorganized hepatic lobular architecture, with widespread hyaline degeneration of hepatocytes (HE staining, 40×/200×). Generated using Adobe Photoshop 2025. NAP-WD: Wilson’s disease without autoimmune phenomena; AP-WD: Wilson’s disease with autoimmune phenomena; HE: hematoxylin and eosin.
Comparison of hepatic histopathological features between the AP-WD group and NAP-WD group
| Variables | NAP-WD group (n = 11) | AP-WD group (n = 6) | Statistic | P |
| AIH-associated histological features | ||||
| Portal interface hepatitis | 0.00 (0.00, 1.00) | 0.50 (0.00, 1.75) | Z = -0.74 | 0.460 |
| Plasma cell infiltration | 0.00 (0.00, 0.00) | 1.00 (0.25, 1.00) | Z = -2.35 | 0.019 |
| Rosette-like structures, n (%) | 1 (9.09) | 3 (50.00) | - | 0.099 |
| General histological features | ||||
| NAS score | 3.10 ± 1.66 | 3.75 ± 2.63 | t = -0.56 | 0.584 |
| Mallory-Denk bodies, n (%) | 1 (9.09) | 1 (16.67) | - | 1.000 |
| The degree of inflammation in the portal areas | 1.00 (1.00, 1.00) | 2.50 (2.00, 3.00) | Z = -2.73 | 0.006 |
| Degree of fibrosis staging, n (%) | - | 1.000 | ||
| S0 | 2 (18.18) | 1 (16.67) | ||
| S1 | 1 (9.09) | 0 (0.00) | ||
| S2 | 4 (36.36) | 2 (33.33) | ||
| S3 | 3 (27.27) | 2 (33.33) | ||
| S4 | 1 (9.09) | 1 (16.67) | ||
Association between AP and LRAEs in WD patients
A total of 66 patients were included in the time-to-event analysis, with follow-up of up to 60 months. During this period, 15 patients experienced LRAEs. In the NAP-WD group, 3 patients experienced LRAEs, all resulting in death. In the AP-WD group, 12 patients experienced LRAEs, including 7 deaths and 5 liver transplantations.
The incidence of LRAEs was significantly higher in the AP-WD group than in the NAP-WD group (P = 0.017) [Figure 4A]. Kaplan-Meier analysis demonstrated a significantly higher cumulative risk of LRAEs in the AP-WD group compared with the NAP-WD group, as assessed by the log-rank test (P = 0.018) [Figure 4B]. Consistently, univariable Cox proportional hazards regression showed that patients in the AP-WD group had a significantly increased risk of LRAEs compared with those in the NAP-WD group [HR (hazard ratio): 3.932, 95%CI: 1.106-13.976].
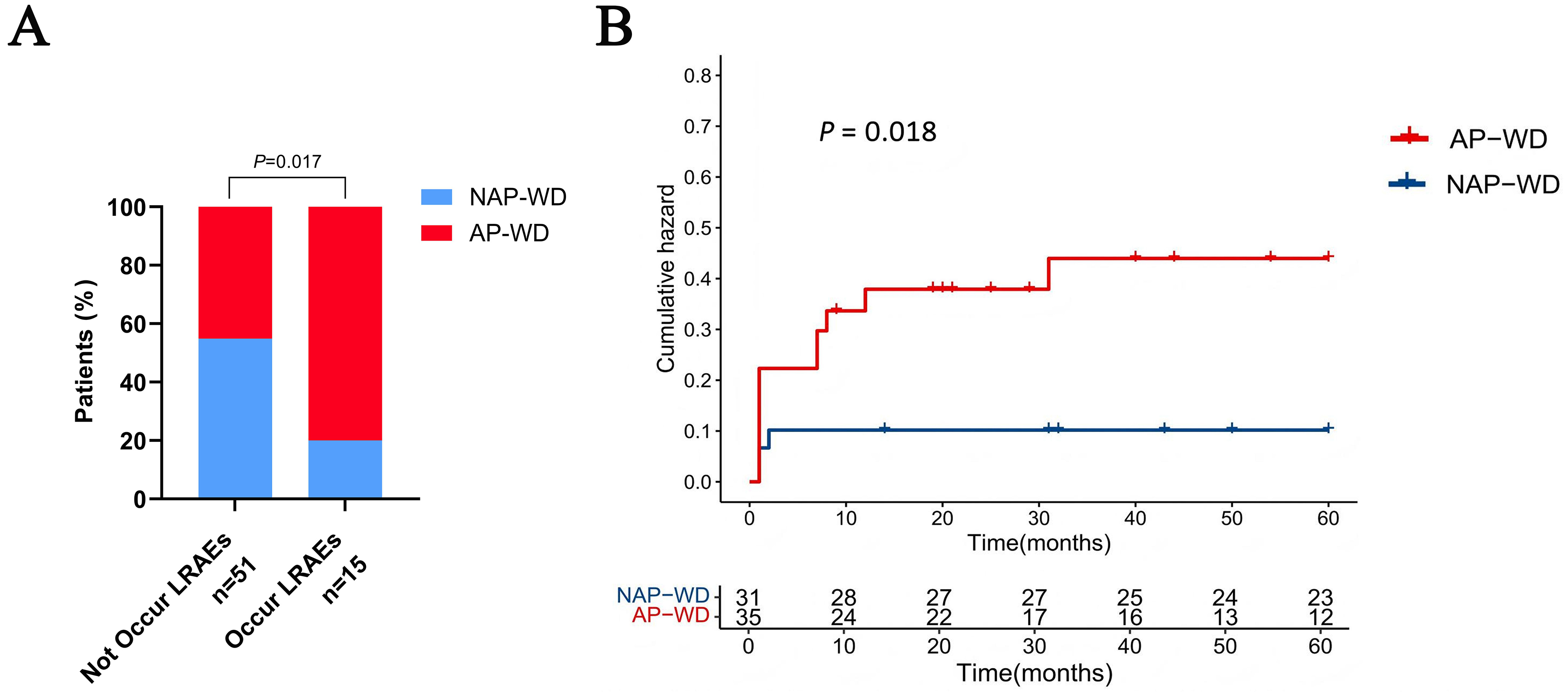
Figure 4. Occurrence and risk of LRAEs in AP-WD and NAP-WD patients. (A) Distribution of patients in the AP-WD and NAP-WD groups according to the presence or absence of LRAEs; (B) Kaplan-Meier curves showing the cumulative risk of LRAEs in the AP-WD and NAP-WD groups. Generated using GraphPad Prism 10 and R 4.3.0. LRAEs: Liver-related adverse events; NAP-WD: Wilson’s disease without autoimmune phenomena; AP-WD: Wilson’s disease with autoimmune phenomena.
DISCUSSION
In this retrospective cohort study, we systematically evaluated the prevalence, clinical significance, and prognostic impact of AP in treatment-naïve WD patients. We systematically evaluated the prevalence, clinical significance, and prognostic relevance of AP, providing detailed cross-sectional and follow-up analyses. Our findings indicate that the presence of AP is associated with more severe hepatic dysfunction at baseline and independently predicts an increased risk of LRAEs during long-term follow-up. These results underscore the clinical importance of assessing autoimmune features in newly diagnosed WD patients for risk stratification and informed management.
First, our study revealed that over half of treatment-naïve WD patients (55.8%) met the predefined criteria for AP, and approximately half (50.6%) exhibited at least one autoantibody, indicating that immune activation is relatively common in treatment-naïve WD patients. This prevalence is consistent with a previous report, which also showed a higher frequency of autoantibody positivity in WD patients compared to the general population[8], suggesting that immune dysregulation may be commonly observed in WD and potentially related to disease pathogenesis. What’s more, ANA was the predominant autoantibody detected, present in nearly all autoantibody-positive patients, with most cases showing isolated ANA positivity. The co-occurrence of other autoantibodies was rare, and the majority of autoantibodies were of low titer, indicating subclinical or early immune activation rather than overt autoimmune disease.
Secondly, our study shows that AP-WD patients exhibit more severe baseline clinical features than those without AP. Specifically, they had lower hemoglobin, red blood cell, and platelet counts; more pronounced coagulation impairment; worse liver function; higher 24 h urinary copper excretion; elevated liver-related scores; and a higher prevalence of cirrhosis, KF rings, and ascites. These findings suggest that autoimmune activation in WD is associated with greater disease severity rather than representing a benign phenomenon. To further substantiate this relationship, we confirmed that immune activation in WD is closely linked to hepatic injury and dysfunction through comprehensive analyses of serum IgG levels. The inverse correlations between serum IgG and hepatic synthetic markers such as albumin and cholinesterase, together with the positive correlation between IgG and INR, indicate that elevated IgG levels reflect impaired hepatic synthetic capacity and coagulation dysfunction. However, Jańczyk et al. reported that in children with WD, ANA positivity showed no significant correlation with transaminase levels, copper metabolism, liver stiffness, or steatosis[8]. This discrepancy may be attributed to differences in study populations and design. Specifically, Jańczyk’s study enrolled pediatric WD patients (mean age 11.6 years), who likely had a shorter disease duration and milder disease severity[8]. In addition, their study did not explicitly exclude patients who had received anti-copper therapy, which can influence immune markers, liver function, and overall disease status, thereby confounding the relationship between baseline immune activation and clinical features. In contrast, the present study specifically focused on treatment-naïve WD patients, predominantly adults, and strictly excluded individuals with prior exposure to anti-copper therapy. This design was intended to minimize treatment-related immunological confounding, allowing a more accurate assessment of the impact of immune dysfunction secondary to copper metabolism disorders on disease status. Overall, these findings imply that evaluating autoimmune status at diagnosis could help identify WD patients at higher risk for severe liver involvement and may aid in optimizing early clinical management.
Next, we further confirmed that immune activation in WD is closely associated with hepatic injury and dysfunction through comprehensive histopathological analyses. Compared with NAP-WD patients, those with AP-WD exhibited more pronounced portal inflammation and greater plasma cell infiltration, features reminiscent of AIH. The enrichment of plasma cells suggests localized intrahepatic antibody production and autoimmune-mediated inflammatory responses directed against self-antigens, linking systemic immune activation to histological liver injury. Moreover, the heightened inflammatory activity in the portal areas of AP-WD patients indicates more severe tissue injury, likely reflecting the synergistic impact of copper accumulation and autoimmune activation. Meanwhile, fibrosis stage and steatosis were similar between groups. Collectively, these findings suggest that immune dysregulation in WD is not merely a secondary consequence of copper toxicity but may be associated with more severe liver injury, although a causal relationship remains to be established.
Lastly, our longitudinal follow-up demonstrated that AP-WD had a significantly higher incidence and cumulative risk of LRAEs compared with NAP-WD. This finding indicates that autoimmune activation in WD is not merely a secondary consequence of copper overload but may be associated with disease progression. In the present retrospective analysis, we did not observe clear differences in initial treatment selection between patients with and without AP, which likely reflects standardized, guideline-driven therapeutic approaches in routine practice[21]. Notably, current international guidelines do not provide specific recommendations for the management of WD patients with concomitant AP, and treatment decisions are therefore largely based on overall disease severity rather than immune features. From our clinical experience, autoimmune manifestations are often interpreted as indicators of more active or advanced disease and may reasonably prompt more aggressive de-coppering strategies and closer monitoring in real-world practice, particularly in patients receiving D-penicillamine, which itself may modulate immune responses. Whether such tailored management improves outcomes in AP-WD patients remains to be determined and warrants prospective validation.
It is worth noting that the clinical manifestations of WD are highly heterogeneous. A previous study[7] has demonstrated that the overall prevalence of conventional autoantibodies does not significantly differ between patients with predominantly neurological (cerebral) phenotypes and those with hepatic phenotypes of WD. Moreover, increasing attention has been paid to neuronal surface antibodies (NSAbs), which have been implicated in cognitive impairment and neurodegenerative disorders, including Alzheimer’s disease, frontotemporal dementia, and amyotrophic lateral sclerosis. Notably, prior reports have shown a higher prevalence of NSAbs in patients with WD compared with healthy controls, whereas no significant differences were observed across WD clinical subtypes or treatment conditions.
Although the pathogenesis of AP in WD remains incompletely defined, several plausible mechanisms may connect chronic copper overload to autoantibody production and immune dysregulation. First, copper toxicity is accompanied by persistent inflammation and oxidative stress, which can promote hepatocyte injury and necrosis, thereby increasing exposure of sequestered self-antigens and facilitating autoantigen availability, particularly in early disease stages[22]. Second, cell and tissue damage generates immune-enhancing “danger” signals, including reactive oxygen species and damage-associated molecular patterns (DAMPs), which can activate sentinel antigen-presenting cells to uptake antigens and migrate to lymphoid tissues for T- and B-cell priming[23]. Third, drawing on general principles of autoimmunity, metals may modify autoantigens and increase their immunogenicity, potentially contributing to a break in immune tolerance through mechanisms such as altered antigen presentation or polyclonal lymphocyte activation[23]. Finally, inflammatory dysregulation involving aberrant T-cell responses (Th1/Th2/Th17) and B-cell activation may further amplify autoantibody generation[24]. These considerations are speculative and hypothesis-generating; dedicated mechanistic studies are required to determine whether and how copper-associated inflammatory injury directly drives AP in WD.
This study has several notable strengths. We established a well-characterized, treatment-naïve cohort, enabling unbiased evaluation of the baseline clinical, immunological, and histopathological features of WD patients. By integrating detailed clinical data, comprehensive serological profiling, and liver histopathology, we provided a multidimensional perspective on the role of AP. Furthermore, long-term follow-up allowed for assessment of liver-related outcomes and evaluation of the prognostic significance of autoimmune markers.
Nevertheless, several limitations should be acknowledged. First, this was a single-center retrospective study with a moderate sample size, which may limit the generalizability of the findings. Second, immune parameters were measured at a single time point, precluding assessment of dynamic changes. Third, mechanistic studies were not performed, and thus the causal link between AP and hepatic injury remains to be elucidated. Future multicenter, prospective studies with longitudinal immune profiling are warranted to validate our findings and clarify the immunopathological mechanisms underlying AP in WD. Fourth, as this study was conducted in a tertiary liver disease referral center, the enrolled cohort predominantly comprised patients with hepatic phenotypes of WD; systematic neurological assessment was not available for most patients. Consequently, the relationship between AP and neurological manifestations could not be comprehensively evaluated. Future multicenter, prospective studies with longitudinal immune profiling and standardized neurological phenotyping are warranted to validate our findings. Fifth, while our study did not identify a clear dose-dependent relationship between autoantibody titers and clinical severity, this observation may in part reflect sample size limitations, particularly in patients with high antibody titers. As a result, subtle titer-related effects cannot be excluded. Future studies with larger cohorts and longitudinal designs will be necessary to determine whether autoantibody magnitude carries independent prognostic significance in WD. Sixth, only 17 treatment-naïve WD patients had available histopathological data. This limited sample size reduces statistical power and raises the possibility of selection bias. Consequently, future studies with larger, prospectively collected biopsy cohorts and standardized histological evaluation will be necessary to confirm and extend these findings. Seventh, to improve comparability between groups, the time-to-event analysis was administratively censored at 60 months[25]. It inevitably limits the assessment of late-occurring LRAEs. As a result, long-term risk beyond the predefined observation window could not be fully evaluated, and the findings should be interpreted within this context. Future studies with larger cohorts and prospectively standardized follow-up durations are warranted.
In conclusion, our study demonstrates that AP is relatively common in WD and is associated with more severe hepatic dysfunction, advanced disease stage, and poorer long-term outcomes. These findings highlight the clinical significance of screening for autoimmune markers in newly diagnosed WD patients and suggest that AP-WD may constitute a distinct clinicopathological subgroup with prognostic relevance. Clinicians should consider incorporating the assessment of autoantibodies and immunoglobulin levels into routine evaluations to identify patients at higher risk of adverse events, thereby enabling tailored monitoring and therapeutic strategies.
DECLARATIONS
Acknowledgment
We thank Ke Li for assistance with Graphical Abstract creation using BioRender.com (Created in BioRender. Jiang, H. (2026) https://BioRender.com/an7sap3). We also thank Manman Xu for her methodological guidance and constructive suggestions during the study design and analysis.
Authors’ contributions
Contribute equally to this work: Jiang H, Tang S
Conceived the study: Jiang H, Zheng S
Interpreted the results and generated the figures and tables: Jiang H, Hou W, Tang S
Collected the data: Jiang H, Liang C, Yu H
Performed liver tissue pathological assessment: Liu H
Carried out telephone follow-up and data entry verification: Zhang S, Li M, Wang Y
Wrote the manuscript: Jiang H, Tang S
Provided feedback and supervised the study: Zheng S, Hou W
Obtained funding and supervised the overall execution of this work: Zheng S
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by grants from the National Key Research and Development Program of Ministry of Science and Technology (2022YFC2304400); High-level public health technical talents of Beijing Municipal Health Commission (Academic Leader -02-14), Beijing Hospitals Authority’s Ascent Plan (DFL20241701), Beijing Science and Technology New Star program cross-cooperation project (20230484455), and the National Natural Science Foundation of China Youth Project Category C (82502232).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
All procedures in this study were conducted in accordance with the ethical standards of the Ethics Committee of Beijing YouAn Hospital, Capital Medical University, and the Declaration of Helsinki (2013 revision). The study was approved by the institutional ethics committee of Beijing YouAn Hospital, Capital Medical University (approval number: LL-2022-048-K). Written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Tang S, Liang C, Yu H, et al. The potential serum sphingolipid biomarkers for distinguishing Wilson disease. Clin Chim Acta. 2024;553:117740.
2. Li S, Lin Y, Chen S, et al. Clinical characteristics and prognosis of early diagnosed Wilson’s disease: a large cohort study. Liver Int. 2024;44:2424-33.
3. Yang RM, Feng T, Cai W, et al. Chinese multidisciplinary expert consensus on orphan/anticopper drugs and other non-drug management of hepatolenticular degeneration. Curr Neuropharmacol. 2025;23:1683-708.
4. Roberts EA, Schilsky ML. Current and emerging issues in Wilson’s disease. N Engl J Med. 2023;389:922-38.
5. Cai H, Cheng X, Wang XP. ATP7B gene therapy of autologous reprogrammed hepatocytes alleviates copper accumulation in a mouse model of Wilson’s disease. Hepatology. 2022;76:1046-57.
6. Brennan PN, Cartlidge P, Manship T, Dillon JF. Guideline review: EASL clinical practice guidelines: drug-induced liver injury (DILI). Frontline Gastroenterol. 2022;13:332-6.
7. Antczak-Kowalska M, Członkowska A, Eyileten C, et al. Autoantibodies in Wilson disease: impact on clinical course. JIMD Rep. 2022;63:508-17.
8. Jańczyk W, Bierła JB, Trojanowska I, Wierzbicka-Rucińska A, Cukrowska B, Socha P. Prevalence and significance of autoantibody seropositivity in children with Wilson’s disease. Diagnostics. 2023;13:768.
9. Santos BC, Guedes LR, Faria LC, Couto CA. Wilson’s disease presentation resembling autoimmune hepatitis. BMJ Case Rep. 2019;12:e230721.
10. Milkiewicz P, Saksena S, Hubscher SG, Elias E. Wilson’s disease with superimposed autoimmune features: report of two cases and review. J Gastroenterol Hepatol. 2000;15:570-4.
11. Lopes SR, Teixeira M, Sequeira C, Carvalho A, Gamito É, Alves AL. Diagnostic pitfalls in Wilson disease with autoimmune features: a case report. GE Port J Gastroenterol. 2025;Epub ahead of print.
12. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139-42.
13. European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69:406-60. (in Chinese).
14. Medical Association Tuberculosis Branch. [Guidelines for diagnosis and management of drug-induced liver injury caused by anti-tuberculosis drugs (2024 version)]. Zhonghua Jie He He Hu Xi Za Zhi. 2024;47:1069-90.
15. Hadjout T, Lamara Mahammed L, Saad M, et al. Is there an alternative to the indirect immunofluorescence ANA HEp-2 assay for the diagnosis of connective tissue diseases? Front Immunol. 2025;16:1669124.
16. Inherited Metabolic Liver Disease Collaboration Group, Chinese Society of Hepatology, Chinese Medical Association. [Guidelines for the diagnosis and treatment of hepatolenticular degeneration (2022 edition)]. Zhonghua Gan Zang Bing Za Zhi. 2022;30:9-20. (in Chinese).
17. Chowdhury AB, Mehta KJ. Liver biopsy for assessment of chronic liver diseases: a synopsis. Clin Exp Med. 2023;23:273-85.
18. Arase Y, Matsumoto K, Anzai K, et al. Clinicopathological features of autoimmune hepatitis with IgG4-positive plasma cell infiltration. Dig Dis. 2021;39:225-33.
19. Rinella ME, Neuschwander-Tetri BA, Siddiqui MS, et al. AASLD Practice Guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology. 2023;77:1797-835.
20. Pashnina IA, Krivolapova IM, Fedotkina TV, et al. Antinuclear autoantibodies in health: autoimmunity is not a synonym of autoimmune disease. Antibodies. 2021;10:9.
21. Association for the Study of the Liver. EASL-ERN Clinical Practice Guidelines on Wilson’s disease. J Hepatol. 2025;Epub ahead of print.
22. Dara N, Imanzadeh F, Sayyari AA, Nasri P, Hosseini AH. Simultaneous presentation of Wilson’s disease and autoimmune hepatitis; a case report and review of literature. Hepat Mon. 2015;15:e29043.
23. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015;125:2228-33.
24. Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001;1:147-53.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.