



Evaluating the prebiotic activity of arabinogalactan on the human gut microbiota using 16S rRNA gene sequencing and Raman-activated cell sorting
 0
0 Abstract
Background: Arabinogalactan is a complex plant-derived polysaccharide proposed to function as a selective prebiotic, yet the microbial taxa directly involved in its metabolism and the cooperative dynamics within the gut microbiota remain incompletely defined.
Methods: Here, we combined community-level sequencing with targeted single-cell activity profiling to investigate how arabinogalactan shapes gut microbial composition and function. Fecal samples from ten healthy individuals were incubated ex vivo with arabinogalactan, and microbial responses were assessed using 16S rRNA gene amplicon sequencing alongside Raman-activated cell sorting (RACS) and coculture experiments.
Results: Arabinogalactan consistently enriched Bifidobacterium and Gemmiger across donors, with Bifidobacterium also responding to galactose and Gemmiger and Blautia stimulated by arabinose, the two monosaccharide components of arabinogalactan. RACS enabled the selective isolation of metabolically active arabinogalactan responders, including Bifidobacterium longum (B. longum) and Faecalibacterium prausnitzii, along with other strains from the phyla Actinomycetota, Bacteroidota, and Bacillota. Notably, coculture experiments revealed that B. longum not only degraded arabinogalactan efficiently but also supported the growth of non-degrading species via metabolic cross-feeding. These cooperative interactions highlight B. longum as a keystone species in arabinogalactan utilization and suggest broader community-level benefits from its activity.
Conclusion: Together, our findings demonstrate arabinogalactan’s bifidogenic effect and its potential to promote functionally important microbes within the gut ecosystem. This study also highlights the utility of RACS for linking microbial identity to function, enabling the targeted recovery of active strains from complex communities.
Keywords
INTRODUCTION
The human gut microbiota is essential for maintaining host health through its roles in nutrient metabolism, immune regulation, and protection against pathogens[1]. Microbial community composition in the gut is influenced by various factors, with diet recognized as a major determinant[2]. At baseline, the adult gut microbiota predominantly consists of bacteria belonging to the phyla Bacillota, Bacteroidota, Actinomycetota, Verrucomicrobiota, and Pseudomonadota[3-5]. However, the relative abundance of these phyla exhibits considerable variability among individuals, underscoring substantial inter-individual differences in microbiota structure and functionality[6]. Non-digestible dietary fibers are increasingly recognized for their ability to selectively modulate the composition and function of the gut microbiota[7]. These fibers, often termed prebiotics, resist digestion by human enzymes, reaching the large intestine intact, where they are selectively fermented by certain gut microbes, thereby conferring health benefits to the host[8]. Well-studied prebiotics include oligosaccharides such as fructooligosaccharides (FOS) and galactooligosaccharides (GOS), as well as polysaccharides like inulin, resistant starch, and beta-glucans, which have been shown to selectively stimulate the growth of beneficial gut microbes such as Bifidobacterium and Lactobacillus[8-10].
Arabinogalactan is a plant-derived polysaccharide that has recently been proposed as a potential prebiotic[11,12]. Prebiotics are defined as substrates selectively utilized by host microorganisms that confer a health benefit[13]. This definition emphasizes both microbial selectivity and demonstrable benefit to the host. In this context, arabinogalactan fulfills key prebiotic criteria by reaching the colon undigested, being selectively fermented by beneficial gut bacteria such as Bifidobacterium, and contributing to the production of health-promoting short-chain fatty acids (SCFAs)[14]. It is naturally abundant in certain dietary sources, notably larch wood and legumes[11,15]. Arabinogalactan is a structurally complex, branched polysaccharide typically composed of a galactan backbone with β-1,3- and/or β-1,6-glycosidic linkages, and contains side chains made of arabinose and galactose residues[16]. Fermentation of arabinogalactan by the gut microbiota results in the production of SCFAs, particularly propionate and butyrate, along with other organic acids[17,18]. These metabolites can contribute to lowering intestinal pH and promoting a gut environment associated with improved host health[19]. Arabinogalactan supplementation has been shown to selectively increase the abundance of Bifidobacterium species, indicating its potential bifidogenic effect[14,20,21]. However, the specific microbial taxa directly involved in arabinogalactan metabolism, as well as the dynamics of their metabolic interactions within the broader microbial ecosystem, remain incompletely characterized[22-25]. Efficient utilization of complex polysaccharides like arabinogalactan can involve cooperative interactions among multiple microbial species[24,26]. Primary degraders can break down complex polysaccharides via CAZymes, releasing metabolites that support secondary consumers[27]. Identifying these microbial interactions is key to understanding how polysaccharides such as arabinogalactan selectively modulate gut microbiota and promote host health[14,27]. These microbial shifts can benefit the host by enhancing SCFA production, supporting gut barrier integrity, and modulating immune responses[28].
To investigate the microbial response to arabinogalactan at both community and single-cell resolution, we employed an integrated ex vivo approach using fecal samples from ten healthy donors. First, we characterized community-level shifts through 16S rRNA gene amplicon sequencing, assessing compositional changes driven by arabinogalactan supplementation. Next, we utilized single-cell Raman microspectroscopy combined with deuterium (D2O) labeling to identify individual microbial cells actively metabolizing arabinogalactan. Metabolically active bacteria were selectively isolated via Raman-activated cell sorting (RACS) and subsequently characterized to confirm their physiological capacity to degrade arabinogalactan. Finally, coculture experiments between a primary arabinogalactan degrader - Bifidobacterium longum (B. longum) - and metabolically active, non-degrading isolates provided insights into potential cooperative interactions, illustrating how B. longum facilitates secondary growth via metabolic cross-feeding. Together, our results provide mechanistic insight into how arabinogalactan selectively enriches specific gut microbes and promotes cooperative interactions, supporting its role as a functionally selective prebiotic.
METHODS
Sample collection and incubation
Stool samples were collected from 10 healthy adults (five males and five females; mean BMI: 24.43 ± 3.69). Participants were excluded if they had taken antibiotics, consumed prebiotic or probiotic products in the past six months, or had a known history of gastrointestinal disorders or recent digestive illness[29,30]. Participants collected their own stool samples using sterile screw-cap containers equipped with built-in collection spoons (Sarstedt, Germany). The study was approved by the Ethics Committee of the University of Vienna, and all individuals provided written informed consent (reference number: 00161).
Immediately following collection, fecal material was transferred into an anaerobic chamber (gas mixture: 85% N2, 10% CO2, 5% H2) to maintain strict anaerobic conditions[31,32]. To assess microbiota responses, fecal homogenates were incubated with arabinogalactan, arabinose, and galactose (2 mg/mL final concentration, Carl Roth and Sigma-Aldrich)[7], while unamended samples were used as negative controls. Samples were homogenized in 2× PBS by vigorous vortexing (2-3 min), passed through a 40 μm mesh filter (Corning, Germany) to remove large debris, and diluted 1:10 with 2× PBS.
Each sugar was dissolved in D2O and added to sterile Hungate tubes containing 2 mL of the fecal homogenate. The final volume was adjusted to 4 mL, yielding a 50% D2O concentration. Incubations were carried out anaerobically at 37 °C for 6 and 24 h. Following incubation, cells were rinsed with PBS to eliminate residual D2O. For downstream processing, 1 mL of each sample was fixed in 50% ethanol/PBS and stored at -20 °C, while an additional 1 mL aliquot was frozen at -80 °C for subsequent nucleic acid and metabolite analysis. For RACS, aliquots were preserved in 20% glycerol/PBS and stored at -80 °C in crimp-sealed vials.
The entire preparation process, including sample homogenization, filtration, and substrate addition, took approximately 30 min. This point was considered the initial time point (0 h) in the dataset and used as the reference for all downstream analyses.
DNA extraction and 16S rRNA amplicon sequencing
To assess shifts in microbial composition, DNA was isolated from stool samples harvested at 0, 6, and 24 h after treatment with arabinogalactan, arabinose, galactose, or without any amendment. DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Germany), incorporating a lysozyme pre-treatment step to enhance cell lysis and optimize DNA recovery[33]. The 16S rRNA gene was amplified using a two-step PCR approach with dual barcoding, allowing for high-resolution profiling of microbial community composition[34]. Sequencing was performed at the Joint Microbiome Facility (JMF) of the Medical University of Vienna and the University of Vienna (Project ID: JMF-2307-05). The resulting sequence datasets have been deposited in the NCBI Short Read Archive under the BioProject accession number PRJNA1244259.
Single-cell Raman analysis of arabinogalactan-stimulated gut microbiota
To assess microbial responses, fecal samples were exposed to arabinogalactan for 6 and 24 h, while parallel incubations without amendment were used as controls. To maintain cellular integrity, samples were fixed in a 1:1 mixture of ethanol and PBS. For Raman measurements, 1 μL of each fixed sample was applied to an aluminum-coated slide (Al136; EMF Corporation, USA) and allowed to air-dry at 30 °C[35,36]. Residual buffer components were removed by dipping the slides twice in ice-cold Milli-Q water (Millipore, Austria), followed by air drying[37].
Spectral acquisition was performed using a confocal Raman microspectroscope (LabRAM HR800, Horiba Scientific, France) equipped with a 532 nm Nd:YAG laser and a diffraction grating of 300 grooves/mm[38]. Prior to measurement, the laser was calibrated using a silicon crystal standard, and the sample slide was brought into focus using a 100× objective[39]. Cell clusters were visually selected, and a mapping grid was applied to the designated area for targeted measurement.
An acquisition time of 0.3 s was chosen to match the conditions used in RACS, allowing direct comparison between the two approaches. Preliminary tests indicated that using 4 accumulations provided the clearest spectra while minimizing laser exposure and preserving cell integrity. A delay time of 0 s was used, and binning was set to 1 to achieve the highest spectral resolution.
A total of 30 to 40 single microbial cells were analyzed per sample. Spectra were considered valid if they showed a clear C–H stretch (2,800-3,100 cm-1) and a phenylalanine peak (~1,000 cm-1), confirming signal origin from cellular material[40,41]. Deuterium incorporation was identified via the C–D stretch (2,040-
The percentage of deuterium-labeled bonds (%CD) was calculated as the ratio of C–D to total C–H and C–D signals using the Scattr analysis tool (https://shiny.lisc.univie.ac.at/scattr/). The threshold for metabolic activity was determined based on %CD values from cells incubated without the addition of D2O (water control), calculating the mean plus 3 times the standard deviation (SD)[38].
Targeted isolation of arabinogalactan-responsive cells via RACS
RACS was performed on fecal samples incubated for 6 h with D2O and arabinogalactan to isolate metabolically active cells. After incubation, cells were preserved in 20% glycerol/PBS and stored at -80 °C. For sorting, frozen samples were thawed, pelleted at 9,000 × g for 7 min, and washed twice with 0.3 M PBS/glycerol solution to minimize osmotic stress[42]. The final cell pellet was resuspended in 500 μL of the same buffer and loaded into a 500 μL Hamilton syringe under anaerobic conditions. The RACS platform consisted of a confocal Raman microscope (532 nm, 90 mW), optical tweezers using a 1,064 nm Nd:YAG laser (500 mW), and a polydimethylsiloxane (PDMS)-based microfluidic sorting device[42]. The PDMS chips were fabricated by mixing base elastomer (Sylgard 184TM, Dow Corning, Michigan, USA) and curing agent at a 10:1 ratio, polymerized at 75 °C, and mounted on a glass coverslip[42]. During the sorting process, cells were sorted by their Raman spectral profiles, allowing deuterium-labeled cells to be directed to the collection outlet, whereas unlabeled cells were guided toward the waste outlet[43]. Cells were identified and sorted based on Raman spectra using automated MATLAB software (version 4.2) based on two indices. The cell index (Pc), which indicate cell capture, was defined as the integrated Raman signal between 1,620-
The sorting process for each sample lasted approximately 60 to 90 min. After sorting, 50 μL of the collected cell suspension was immediately plated on YCFA agar plates and incubated anaerobically at 37 °C. A 50 μL aliquot of the sheath buffer was also plated as a negative control. Colonies recovered after initial plating were subsequently transferred onto YCFA-arabinogalactan plates for confirmation of growth, and successfully grown isolates were preserved as glycerol stocks at -80 °C. The RACS platform has an average throughput of up to 500 cells per hour and a sorting accuracy of 98.3% ± 1.7%[43]. For each donor, the total number of D2O-labeled cells analyzed, sorted, and successfully cultivated was recorded. The post-sorting cultivation success rate, defined as the percentage of colonies recovered relative to the number of labeled cells sorted, ranged from 0.6% to 32.96% across donors [Supplementary Table 1].
16S rRNA gene sequencing of RACS-isolated bacteria
To genetically characterize the bacterial isolates obtained from RACS, colony PCR targeting the 16S rRNA gene was conducted. All PCR mixtures were assembled in a sterile PCR hood to prevent contamination. The reaction mix (50 μL total volume) contained the following components: 5 μL of 10× Green Dream Taq Buffer with 20 mM MgCl2 (Thermo Fisher Scientific, USA), 5 μL of 2 mM dNTP mix (Thermo Fisher Scientific), 1 μL of each primer, 616V (5′-AGA GTT TGA TYM TGG CTC AG-3′) and 1492R (5′-GGT TAC CTT GTT ACG ACT T-3′) with the final concentration of 50 μM (Thermo Fisher Scientific),
Growth curve analysis
Bacterial strains identified by Sanger sequencing were grown anaerobically at 37 °C in both solid and liquid YCFA-glucose (YCFA-G) medium. Once cultures reached the early stationary phase, as determined by OD600, cells were harvested, washed, and resuspended in fresh YCFA medium containing either arabinogalactan (YCFA-AG) or no supplement (YCFA-NA) as a control. Each culture was distributed into a sterile 96-well microplate (Costar 3595, Corning, NY, USA) in triplicate. The plate was incubated for 48 h at 37 °C using an anaerobic microplate reader (MultiskanTM GO, Thermo Fisher Scientific), with optical density at 600 nm measured every 30 min and continuous shaking (5 s before each readout) controlled by SkanIt Software RE (v6.1.0.51). Growth kinetics were analyzed using R (https://www.r-project.org/), and the resulting culture supernatants were harvested and stored at -80 °C.
Coculture evaluation
To investigate potential metabolic interactions, coculture experiments were performed using B. longum and eight non-degrading strains previously identified via RACS as unable to utilize arabinogalactan independently. These strains included E. lenta, C. aerofaciens, P. coprocola, D. welbionis, R. bicirculans,
Strain-specific primers were designed using Primer3Plus to enable quantitative PCR (qPCR) analysis of strain abundance in both monocultures and cocultures[45]. The primers used for each strain were as follows: BifL-F (GAGATACGGCTTCCCTTCGG) and BifL-R (CATAATCCGCTGGCAACACG) for B. longum (Product length: 126 bp, Tm: 60 °C), Egg-F (CGCGGCCCATTAGGTAGTAG) and Egg-R (AGTCTGGGCCGTATCTCAGT) for E. lenta (Product length: 106 bp, Tm: 60 °C), Coll-F (TGCTACAATGGCCGGTACAG) and Coll-R (AGCAACTCCGACTTCATGGG) for C. aerofaciens (Product length: 114 bp, Tm: 60 °C), Phoc-F (CGTGAGGTGTCGGCTTAAGT) and Phoc-R (TCCTCGCATCTTACGATGGC) for P. coprocola (Product length: 105 bp, Tm: 60 °C), Dyso-F (GAGCTCGCGTCTGATTAGCT) and Dyso-R (TGTCTCAGTCCCAATGTGGC) for D. welbionis (Product length: 100 bp, Tm: 60 °C), Rumino-F (GAGCTCGCGTCTGATTAGCT) and Rumino-R (TGTCTCAGTCCCAATGTGGC) for R. bicirculans (Product length: 100 bp, Tm: 60 °C), Phas-F (AGTAAACGAGGAAGCCACGG) and Phas-R (AAGCCGCCTACATGCTCTTT) for P. faecium (Product length: 105 bp, Tm: 60 °C), Para-F (GTGTGTTTGAGGTAGGCGGA) and Para-R (GTAAGCTGCCTTCGCAATCG) for P. merdae (Product length: 84 bp, Tm: 60 °C), Alis-F (AGCTGGTTGGTGAGGTAACG) and Alis-R (TGGTCCGTGTCTCAGTACCA) for A. shahii (Product length: 89 bp, Tm: 60 °C). Primer specificity was validated using PCR with both specific and non-specific templates and optimized using gradient PCR to determine optimal annealing temperatures. For qPCR quantification, DNA standards were prepared by growing each strain to OD600 = 0.1, followed by DNA extraction. The 10-fold serial dilutions of the extracted DNA were used to establish standard curves for absolute quantification. The qPCR reactions were run in triplicate for each sample, enabling accurate quantification of bacterial abundance in both monocultures and cocultures across the selected time points. This setup allowed us to evaluate strain-specific growth responses and potential cross-feeding interactions mediated by B. longum during arabinogalactan degradation.
Statistical analysis and reproducibility
All analyses were conducted in R statistical software (v4.2.1; https://www.r-project.org/)[46]. Data preprocessing and transformation were performed using the data.table, dplyr, and tidyr packages, and visualizations were generated with ggplot2 (v3.5.1)[47]. Microbial community-level differences were assessed using the vegan package (v2.6.4)[48], applying Bray–Curtis dissimilarity and permutational multivariate analysis of variance (PERMANOVA)[48]. To evaluate taxon-level responses to dietary amendments, enrichment factors (EFs) were calculated by comparing genus-level relative abundances at 6 and 24 h to their corresponding baseline [no amendment (NA)] at the same time points. The EF was computed using the formula EF = 2A/(A + B) - 1, where A and B represent the relative abundances in treated and control conditions, respectively. This approach enabled genus-level comparisons across donors while keeping the output values within a defined and symmetric range between -1 and 1. Because we did not include external spike-ins or perform absolute quantification, EFs were calculated based on relative changes within each individual donor, comparing treated and control conditions separately for each donor. Genera with EF > 0 were considered enriched, and those with EF < 0 depleted. Statistical significance was assessed using z-scores derived from RA comparisons, with P-values adjusted using the Benjamini-Hochberg false discovery rate (FDR) method[49]. Genera with EF > 0 and FDR-adjusted P < 0.05 were considered significantly enriched. Bubble plots were used to visualize results, where bubble size reflected the relative abundance of baseline at the initial time point (0 h), bubble color encoded EF, and statistically significant genera were annotated with asterisks. DESeq2 (v1.38.3) was also used for differential abundance testing, modeling genus-level count data with a negative binomial distribution[50]. Donor identity and time point were included as variables in the model design. We selected DESeq2 for this analysis because it supports modeling donor identity and treatment effects, which was essential for accounting for the high inter-individual variability observed across donors. Genera with FDR-adjusted P < 0.05 were considered significantly differentially abundant. The combination of EF and DESeq2 provided complementary insights into both donor-specific and consistent taxon-level responses to arabinogalactan supplementation. To assess shifts in microbial community structure between arabinogalactan-amended and control samples, PERMANOVA was performed using the vegan package (v2.6.4) in R. The adonis function was applied to test for differences in community composition based on Bray–Curtis dissimilarity metrics computed from genus-level relative abundance profiles[48]. This multivariate approach allowed us to evaluate whether overall microbial community structures varied significantly between treatments, beyond changes at the individual taxon level.
RESULTS
Selective enrichment of gut microbes in response to arabinogalactan supplementation
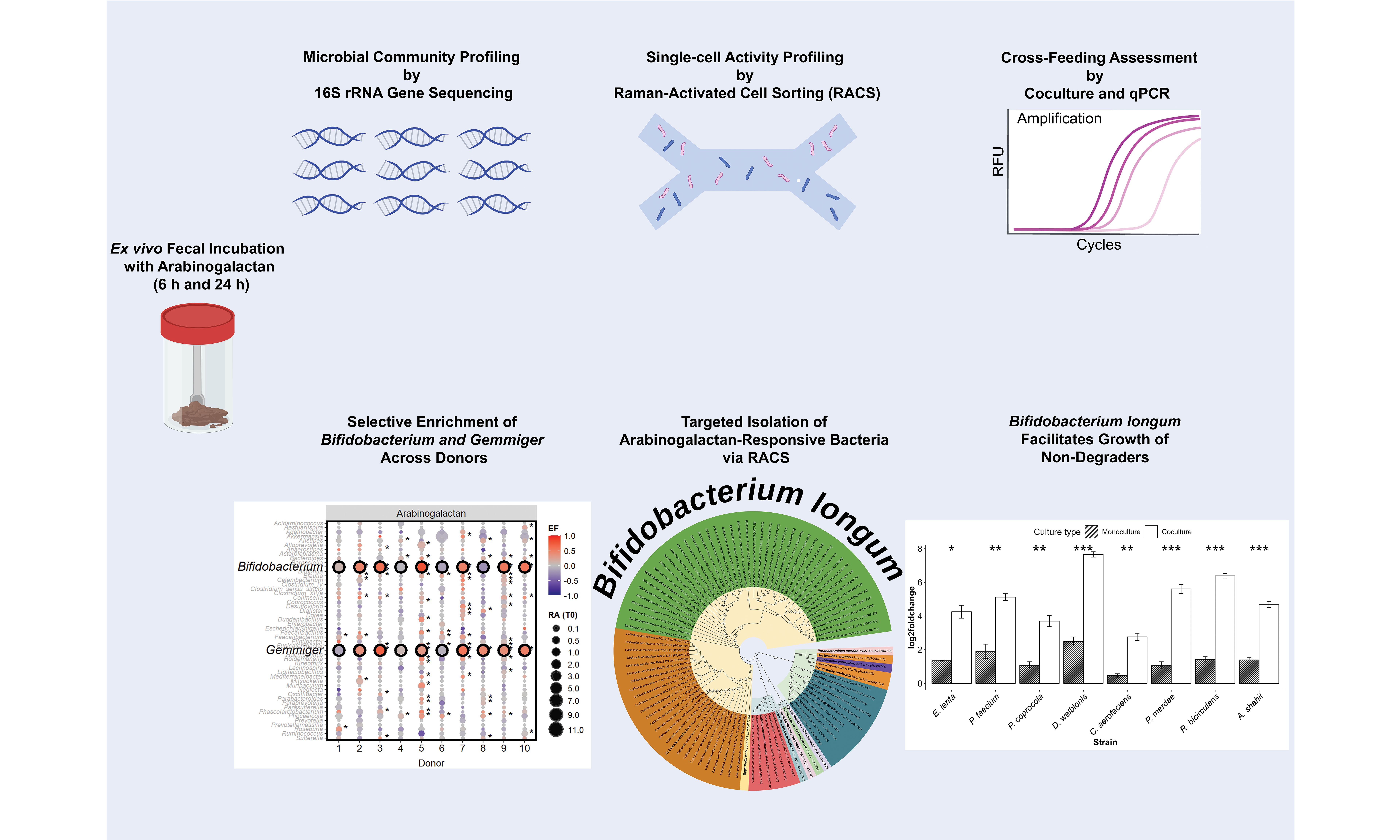
To characterize the baseline microbial community structure prior to incubation, we performed 16S rRNA gene amplicon sequencing on fecal samples collected from all ten donors at the initial time point (0 h). Across the donors, the gut microbiota was predominantly composed of members of the phyla Bacillota (50.8%) and Bacteroidota (40.3%), with lower contributions from Verrucomicrobiota (4.1%), Pseudomonadota (2.9%), and Actinomycetota (1.9%) [Figure 1A].
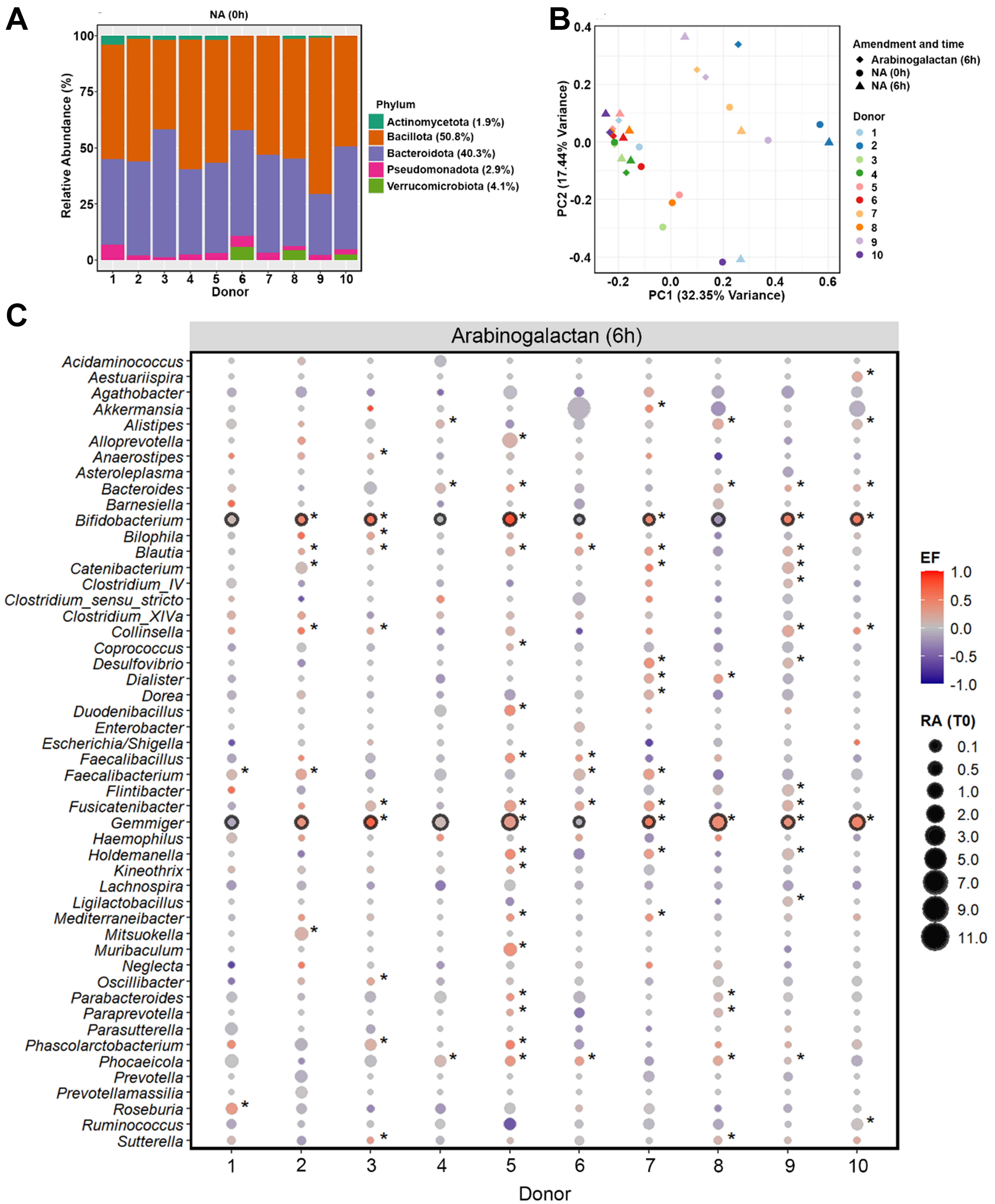
Figure 1. Baseline gut microbiota composition and response to arabinogalactan. (A) Relative abundances (%) of fecal microbiota at the phylum level across ten donors at the baseline (0 h). Average values for each phylum across all donors are provided in the legend; (B) PCoA of genus-level microbial communities, illustrating the clustering pattern of samples after 6-hour incubation with arabinogalactan, compared to NA samples at 0 and 6 h. Color and shape indicate donor identity and treatment condition, highlighting both individual and treatment-related differences; (C) Enrichment patterns of dominant bacterial genera after 6 h incubation with arabinogalactan. Bubble size indicates the relative abundance at 0 h, and color indicates the scaled EF, as described in the METHODS section. Genera with significant enrichment in individual donors are indicated by an asterisk, while those consistently enriched across all donors are highlighted with a black outline (DESeq2, Wald Test P.adj < 0.05, n = 30 samples). PCoA: Principal coordinate analysis; NA: no amendment; EF: enrichment factor.
Analysis of genus-level microbiome profiles revealed that most of the variation in microbial composition was explained by donor-specific differences, with inter-individual effects accounting for 71.3% of the total variation (R2 = 0.713, P = 0.001; n = 20 samples). This donor effect was also the main factor driving the clustering pattern observed in the principal coordinates analysis, indicating pronounced baseline differences in gut microbiota across individuals [Figure 1B]. Despite this variation, microbial communities incubated with arabinogalactan for 6 h showed a consistent shift relative to 0 and 6 h NA samples. A significant effect of arabinogalactan treatment on microbial community composition was detected (R2 = 0.121, P = 0.004), indicating that the induced shifts were consistently reproducible across distinct microbiota backgrounds.
To assess the extended short-term effects of supplementation, we incubated samples for 24 h. Similar to the 6 h time point, community composition remained strongly donor-driven (R2 = 0.703, P = 0.001), yet arabinogalactan treatment still explained a significant portion of the variation between groups (R2 = 0.123, P = 0.004). These results confirm that the observed community shifts are sustained over time and continue to reflect a robust and reproducible response to arabinogalactan across different individuals [Supplementary Figure 1].
We next identified bacterial genera that were selectively enriched in response to arabinogalactan after 6 h of incubation. Genus-level differential abundance analysis revealed that Bifidobacterium and Gemmiger showed a consistent and significant increase across the donors, but not in the NA samples (DESeq2, Wald Test P.adj < 0.05; Figure 1C, Supplementary Figure 2). This highlights a conserved response to arabinogalactan supplementation, suggesting these genera may play a central role in its metabolism. The enrichment of Bifidobacterium across the donor samples reflects a robust bifidogenic response to arabinogalactan treatment. This effect was not observed in unamended controls, highlighting that the enrichment was specifically induced by arabinogalactan. In addition, other taxa - including Alistipes, Bacteroides, Blautia, Catenibacterium, Collinsella, Faecalibacterium, Oscillibacter, Parabacteroides, Phascolarctobacterium, Phocaeicola, and Ruminococcus - showed significant enrichment in one or more donors, reflecting donor-specific shifts in microbial composition [Supplementary Data 1].
To assess whether these effects were maintained or intensified over time, we performed an additional incubation with arabinogalactan for 24 h. The enrichment patterns observed at 6 h were largely preserved, with Bifidobacterium and Gemmiger continuing to show significant increases across the donors
To further distinguish the effects of arabinogalactan from those of its constituent monosaccharides, we incubated the same donor samples with either galactose or arabinose for 6 and 24 h. Galactose consistently stimulated a significant enrichment of Bifidobacterium across the donors at both time points
Targeted cultivation of arabinogalactan-responsive bacteria using Raman-based activity profiling
To identify metabolically active gut microbes responding to arabinogalactan, we performed D2O labeling followed by single-cell Raman microspectroscopy. Deuterium incorporation, quantified as the percentage of C–D bonds relative to total C–H and C–D (%CD), was used to assess microbial metabolic activity[35]. After
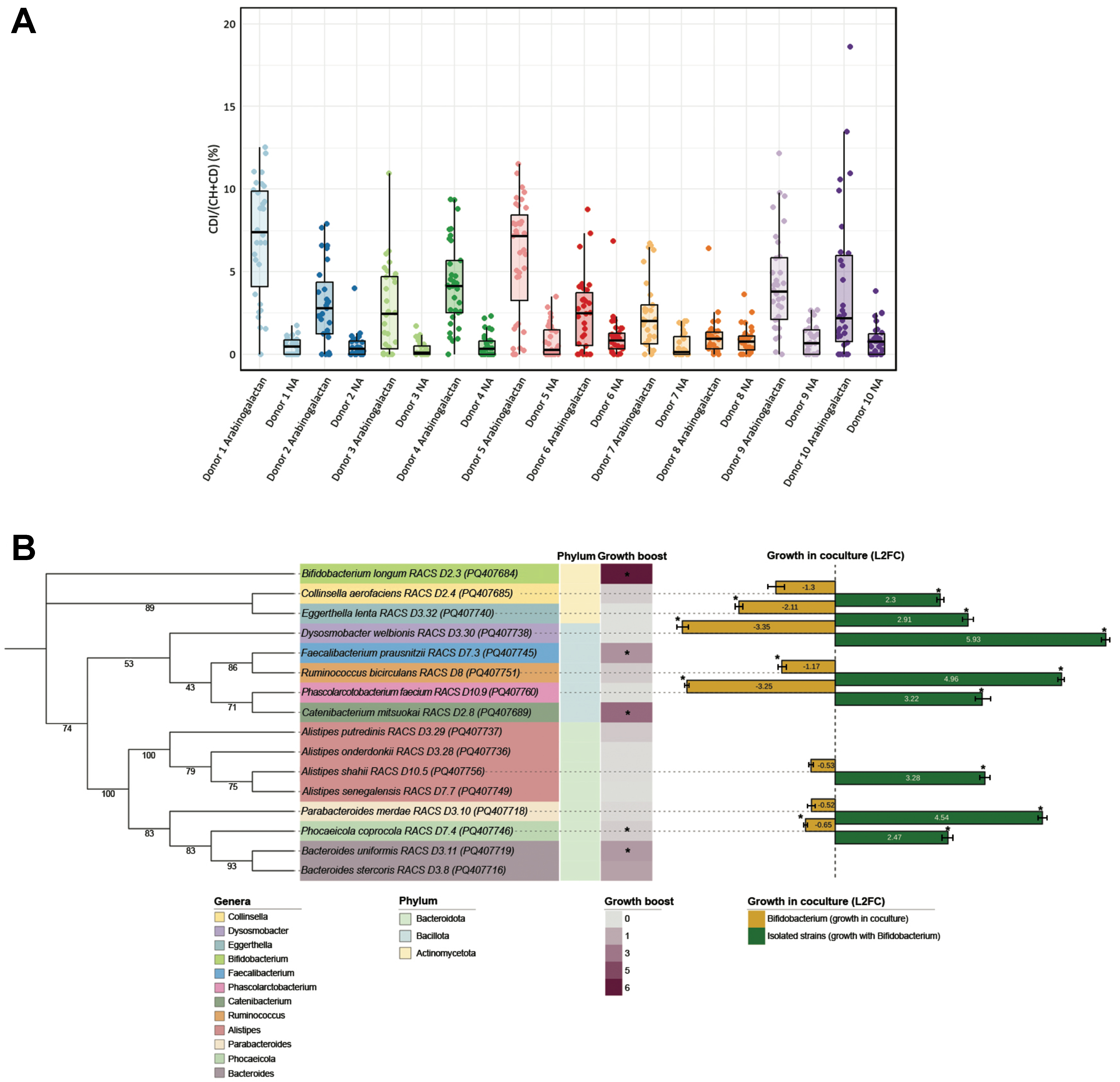
Figure 2. Raman-based detection and physiological analysis of arabinogalactan-responsive gut microbes. (A) Single-cell analysis of microbial metabolic activity in response to arabinogalactan. The percentage of deuterium incorporation (%CD) was measured in individual cells following 6 h of incubation with D2O, with or without arabinogalactan. The dots indicate single-cell measurements collected from each ten donors, while boxplots illustrate the spread of %CD values across conditions. Statistical testing across all donors revealed a significant increase in microbial metabolic activity following arabinogalactan treatment (ANOVA, P < 0.001, n = 607); (B) Phylogenetic and physiological profiling of representative arabinogalactan-responsive isolates. A mid-point rooted maximum likelihood phylogenetic tree was generated from near full-length 16S rRNA gene sequences of 16 representative strains isolated via RACS. Branch support values are based on 1,000 ultrafast bootstrap replicates. The tree generation was performed using the aligned sequences, with subsequent visualization annotated in iTOL[51]. The heatmap displays growth boost, calculated as the area under the growth curve in arabinogalactan-supplemented media relative to no-amendment control, based on three technical replicates per condition. Asterisks indicate statistically significant differences in AUC between the two conditions (Student’s t-test, P < 0.05, n = 6). Color. Bar plots show L2FC in abundance of each strain after 24 h of coculture with B. longum, as quantified by qPCR based on the average of three technical replicates. Asterisks (*) indicate significant differences in growth (Student’s t-test, P < 0.05, n = 6). ANOVA: Analysis of variance; RACS: Raman-activated cell sorting; iTOL: interactive tree of life; AUC: area under the curve; L2FC: log2 fold change; qPCR: quantitative PCR.
To isolate metabolically active taxa responsive to arabinogalactan, we next employed RACS following 6 h of incubation with D2O and arabinogalactan. A total of 98 strains were recovered from RACS-enriched fractions across all donors and classified via near-full-length 16S rRNA gene sequencing as Bifidobacterium longum (n = 45), Collinsella aerofaciens (n = 26), Alistipes shahii (n = 5), Alistipes senegalensis (n = 1), Alistipes putredinis (n = 2), Alistipes onderdonkii (n = 3), Bacteroides uniformis (n = 2), Bacteroides stercoris
A total of 16 strains were selected as representative isolates for downstream analysis [Figure 2B]. One representative per species was chosen to assess its physiological response to arabinogalactan. To functionally characterize arabinogalactan responders, we selected 16 representative isolates from the 98 strains recovered by RACS, ensuring broad phylogenetic coverage while minimizing redundancy among closely related strains. A single B. longum strain was chosen for coculture assays based on its high recovery frequency (n = 45) and robust growth on arabinogalactan. Growth assays and coculture experiments were conducted in triplicate. Quantitative growth boosts were calculated as area under the curve (AUC) in arabinogalactan-supplemented versus no-amendment media, with statistical significance determined by Student’s t-test (P < 0.05). The log2 fold changes in coculture abundance, quantified by qPCR, represent mean ± SD from three replicates. Error bars and individual data points are included to illustrate replicate variation. The addition of arabinogalactan to the cultivation medium stimulated the growth of B. longum, F. prausnitzii, C. mitsuokai, B. uniformis, and B. stercoris.
Since the remaining strains had incorporated deuterium from D2O (indicating metabolic activity) but were unable to utilize arabinogalactan as a sole carbon source, we hypothesized that their growth might be indirectly supported by primary arabinogalactan degraders. B. longum was selected as the focal degrader as it was the most frequently isolated species and exhibited robust growth on arabinogalactan. To test this hypothesis, we performed coculture experiments in which each non-arabinogalactan-utilizing isolate was incubated with B. longum for 24 h in the presence of arabinogalactan. As expected, none of these strains grew in monoculture, but all displayed significant growth when cocultured with B. longum, suggesting that B. longum facilitated their proliferation through metabolic cross-feeding. Notably, the growth of B. longum itself was significantly reduced in cocultures with Eggerthella lenta, Dysosmobacter welbionis, Ruminococcus bicirculans, Phascolarctobacterium faecium, and Phocaeicola coprocola.
DISCUSSION
Dietary fibers that selectively stimulate specific gut microbes are increasingly recognized for their potential to promote host health by modulating immunity, improving gut function, and contributing to the production of bioactive metabolites such as SCFAs[7,13,52,53]. Prebiotics such as inulin, FOS, and GOS have been explored for their capacity to selectively enrich microbial groups involved in carbohydrate fermentation and SCFA production[8,38]. These include Bifidobacterium, Lactobacillus, and Faecalibacterium, which are frequently associated with gut health and beneficial physiological functions[38,54-57]. However, their effects are not always consistent across studies, and the underlying microbial mechanisms are still not fully understood[58,59]. Bifidobacterium, in particular, is widely recognized as a keystone member of the gut microbiota with a central role in fiber degradation and ecosystem function[60].
In this study, we evaluated the selective effects of arabinogalactan, a complex polysaccharide composed of the monosaccharides galactose and arabinose. Arabinogalactan consistently enriched both Bifidobacterium and Gemmiger across the donors at both the 6 and 24 h incubation time points. However, this pattern was not observed in unamended controls. Interestingly, galactose alone led to a consistent enrichment of Bifidobacterium, while arabinose selectively enriched Gemmiger at 6 h and Blautia and Anaerostipes at 24 h. These findings show that while each monosaccharide selectively stimulated specific taxa, the intact polysaccharide elicited a more coordinated response, simultaneously promoting both Bifidobacterium and Gemmiger in a way that was not observed with the individual sugars. Notably, similar enrichment patterns in response to galactose, arabinose, and arabinogalactan have been reported in previous studies and are consistent with our findings[60-65]. For instance, the enrichment of Bifidobacterium in response to galactose has been reported in transcriptomic studies examining growth on substrates rich in GOS and galactose[61]. In another study, Blautia proliferation increased via cross-feeding, following the release of arabinose from arabinoglycan-containing diets in the gut of malnourished mice[60]. Additionally, B. longum was found to be stimulated during the in vitro fermentation of arabinogalactan using a dynamic colon model[65]. These overlaps highlight the consistency of certain microbial responses, while our use of RACS-based isolation and strain-level functional validation provides new insights into active degraders and cooperative interactions, distinguishing our study from previous work.
B. longum was identified as a key species in arabinogalactan degradation, both in terms of abundance and metabolic function. It was the most frequently recovered taxon using RACS and demonstrated the ability to degrade arabinogalactan as a sole carbon source. Additional strains such as F. prausnitzii, C. mitsuokai,
Similar to Bifidobacterium, Gemmiger displayed a robust increase across the donor samples following arabinogalactan supplementation. However, despite its consistent enrichment, Gemmiger was not among the metabolically active strains isolated using RACS. This may be due to cultivation challenges or limitations in cell sorting, possibly related to its strict growth requirements[72-74]. While RACS is highly effective for detecting and isolating metabolically active cells, the technique has certain limitations. The laser used for Raman spectroscopy can transfer energy that may damage fragile cells and reduce their viability. Additionally, post-sorting recovery can be challenging for strictly anaerobic taxa that are highly sensitive to oxygen or require very specific growth conditions, which can limit successful cultivation after sorting. Interestingly, we successfully isolated F. prausnitzii, an oxygen-sensitive butyrate producer closely related to Gemmiger[75]. A 16S rRNA sequence comparison revealed that the dominant Gemmiger ASV shares 92.79% identity with Faecalibacterium, suggesting phylogenetic proximity and possibly overlapping ecological roles[76,77]. This suggests that both taxa may participate in functions such as arabinogalactan fermentation and butyrate production under anaerobic conditions. However, individual strains can differ in which sugars they break down, which enzymes they produce, and how well they grow under gut conditions. Therefore, detailed genome sequencing and experimental validation of substrate utilization and enzyme expression will be important to determine whether Gemmiger functionally overlaps with F. prausnitzii or contributes unique metabolic activities within the gut community.
Our findings highlight arabinogalactan as a functionally selective prebiotic that enriches specific gut microbes, most notably B. longum. Unlike other confirmed prebiotics such as inulin or GOS, which stimulate multiple primary degraders[8,38,78], arabinogalactan targets a narrower group of responsive taxa.
While 16S rRNA gene sequencing provided valuable insights into community-level shifts in response to arabinogalactan, it has notable limitations in taxonomic resolution, functional interpretation, and differential abundance testing. For instance, the 16S rRNA gene cannot reliably distinguish closely related species or infer the enzymatic capabilities responsible for fiber degradation. To address these challenges, we used RACS to isolate and validate metabolically active taxa at the strain level. This dual approach helped bridge the gap between community profiling and functional activity. However, future studies could benefit from incorporating multi-omics strategies such as shotgun metagenomics, metatranscriptomics, or metabolomics to resolve microbial interaction networks, uncover the genetic basis of arabinogalactan metabolism, and characterize host-relevant metabolic outputs with greater phylogenetic and functional resolution. In future studies, using absolute microbiome quantification approaches such as those based on internal standards or spike-ins could provide more accurate estimates of microbial abundance. Future work should clarify the metabolic pathways involved in arabinogalactan degradation, including the identity of breakdown products and their role in supporting non-degraders. Metabolite profiling of B. longum could reveal key compounds responsible for these effects. Additionally, profiling the metabolites produced by
There are several limitations to this study that should be acknowledged. First, our reliance on 16S rRNA gene sequencing restricts taxonomic resolution and does not provide direct information on the functional activities, metabolic pathways, or specific enzymes involved in arabinogalactan metabolism. However, this approach still allowed us to robustly identify key taxa that responded to arabinogalactan, providing valuable ecological insights. This limits our ability to draw conclusions about the metabolic mechanisms driving the observed microbial changes. In addition, we did not perform direct enzymatic assays, which would have helped confirm the involvement of particular enzymes in arabinogalactan degradation. All data are reported as relative abundances rather than absolute quantities. Implementing absolute quantification methods, such as digital PCR (dPCR) or spike-in standards, in future studies could enable more robust and quantitative assessments of microbial dynamics[86]. Despite this, the changes in relative abundance provided consistent and interpretable patterns across donors. The RACS technique, while powerful for isolating metabolically active cells, also presents challenges. Some taxa, such as Gemmiger, were not recovered, likely due to cultivation difficulties, sensitivity to laser exposure, or because strictly anaerobic organisms are more difficult to isolate with this method. As a result, some metabolically active and enriched taxa may have been missed among the isolates. Improved cultivation protocols and alternative single-cell approaches could help address this limitation. Furthermore, while coculture experiments indicated cross-feeding interactions, the precise metabolic intermediates exchanged between strains were not identified. Targeted metabolomics and comprehensive analysis of arabinogalactan utilization would be valuable for elucidating the nature of these metabolic exchanges and utilization patterns. Nonetheless, our experiments demonstrated clear ecological relationships and potential for cooperative interactions within the microbial community. Our study was conducted in vitro using fecal samples from healthy donors, but the effects of arabinogalactan on microbiota from individuals with dysbiosis or gut disease remain unexplored. Another limitation is the limited number of human donors included in this study. A larger number of donors and samples would make the results more reliable and improve our findings across more diverse populations. Future research involving clinical studies or animal models will be important to assess the impact of arabinogalactan supplementation under more physiologically relevant and disease-specific conditions. Finally, the absence of complementary multi-omics approaches, such as shotgun metagenomics, transcriptomics, or genome sequencing, limits the functional interpretation of our findings. Integrating these techniques in future research will be essential for a more comprehensive understanding of arabinogalactan’s impact on the gut microbiome. Despite these limitations, the integrative methods used in this study provide a strong foundation for future investigations.
In conclusion, this study demonstrates that arabinogalactan acts as a functionally selective dietary fiber that consistently enriches beneficial members of the gut microbiota, most notably B. longum. Using an integrated ex vivo framework combining 16S rRNA gene sequencing with RACS, we identified key taxa actively responding to arabinogalactan supplementation across microbiota from multiple human donors. Among the 98 metabolically active strains isolated, B. longum accounted for approximately 46% and was selected for detailed physiological characterization based on its high recovery rate and known role in fiber degradation. B. longum emerged as a central degrader, directly utilizing arabinogalactan and facilitating the growth of non-degrading strains through metabolic cross-feeding. While isolates from Bacteroidota and Bacillota were also recovered, this study focused on Actinomycetota due to the dominance of Bifidobacterium in the RACS-sorted fraction. The targeted recovery and physiological assessment of isolates from three dominant gut phyla Actinomycetota, Bacteroidota, and Bacillota revealed that most metabolically active strains belonged to Actinomycetota, primarily due to the high prevalence of Bifidobacterium. These results underscore arabinogalactan’s selective influence on gut microbial community composition and highlight the potential of single-cell techniques like RACS to uncover functionally important microbial interactions within complex ecosystems.
DECLARATIONS
Authors’ contributions
Planned and designed the study: Rasoulimehrabani H, Berry D
Conducted all the experiments and performed the data analyses: Rasoulimehrabani H
Performed the RACS experiments and contributed data and bioinformatic analysis: Khadem S
Conducted the Raman microspectroscopy measurements: Philipp M
Contributed to molecular biology experiments: Nikolov G
Designed and supported the coculture experiments: Hodžić A
Carried out sample collection and incubation: Khadem S, Gallo R
Contributed to bioinformatic processing: Séneca J
Extracted DNA from all samples: Ramesmayer J
Contributed to the growth curve and coculture experiments: Sivulič P
Wrote the manuscript with input from all co-authors: Rasoulimehrabani H, Berry D
All authors approved the final version of the manuscript.
Availability of data and materials
All 16S rRNA gene amplicon sequencing data generated during this study are available in the NCBI Short Read Archive under the accession number PRJNA1244259. The sequences of RACS-isolated strains following arabinogalactan supplementation have been submitted to GenBank under accession numbers PQ407681 to PQ407778. The Raman spectroscopy dataset has been submitted to the BioStudies database and is accessible through the MicrobioRaman platform under the accession number S-MBRS14.
Financial support and sponsorship
This study received financial support from the European Research Council (FunKeyGut 741623) and the Austrian Science Fund (10.55776/DOC69; 10.55776/COE7).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
All experimental procedures involving human fecal samples were conducted in accordance with the approval of the Ethics Committee of the University of Vienna (reference number: 00161), and all participants provided written informed consent.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
2. David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559-63.
3. Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480-4.
4. Qin J, Li R, Raes J, et al; MetaHIT Consortium. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59-65.
5. Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635-8.
6. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207-14.
8. Gibson GR, Hutkins R, Sanders ME, et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroenterol Hepatol. 2017;14:491-502.
9. Davani-Davari D, Negahdaripour M, Karimzadeh I, et al. Prebiotics: definition, types, sources, mechanisms, and clinical applications. Foods. 2019;8:92.
10. Holscher HD. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes. 2017;8:172-84.
11. Dion C, Chappuis E, Ripoll C. Does larch arabinogalactan enhance immune function? A review of mechanistic and clinical trials. Nutr Metab. 2016;13:28.
12. Uauy R. The assessment of dietary adequacy based on nutrient intake data is a complex issue. Foreword. Br J Nutr. 2009;101 Suppl 2:S1.
13. Gibson GR, Roberfroid MB. Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr. 1995;125:1401-12.
14. Wang Y, LaPointe G. Arabinogalactan utilization by Bifidobacterium longum subsp. longum NCC 2705 and Bacteroides caccae ATCC 43185 in monoculture and coculture. Microorganisms. 2020;8:1703.
15. Kim K, Kim CY. Rheological properties of arabinogalactan solutions related to the carbohydrate composition of different legumes. Korean J Food Preserv. 2023;30:785-96.
16. Rakhmanberdyeva RK, Zhauynbayeva KS, Senchenkova SN, Shashkov AS, Bobakulov KM. Structure of arabinogalactan and pectin from the Silybum marianum. Carbohydr Res. 2019;485:107797.
17. Wang M, Wichienchot S, He X, Fu X, Huang Q, Zhang B.
18. Sun Y, Hu J, Zhang S, et al. Prebiotic characteristics of arabinogalactans during in vitro fermentation through multi-omics analysis. Food Chem Toxicol. 2021;156:112522.
19. Victoria Obayomi O, Folakemi Olaniran A, Olugbemiga Owa S. Unveiling the role of functional foods with emphasis on prebiotics and probiotics in human health: a review. J Funct Foods. 2024;119:106337.
20. Wang Y, Liu Y, Ivusic Polic I, Chandran Matheyambath A, Lapointe G. Modulation of human gut microbiota composition and metabolites by arabinogalactan and Bifidobacterium longum subsp. longum BB536 in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME®). J Funct Foods. 2021;87:104820.
21. Sasaki Y, Horigome A, Odamaki T, et al. Novel 3-O-α-D-Galactosyl-α-L-arabinofuranosidase for the assimilation of gum arabic arabinogalactan protein in Bifidobacterium longum subsp. longum. Appl Environ Microbiol. 2021;87:e02690-20.
22. Onumpai C, Kolida S, Bonnin E, Rastall RA. Microbial utilization and selectivity of pectin fractions with various structures. Appl Environ Microbiol. 2011;77:5747-54.
23. Lindstad LJ, Lo G, Leivers S, et al. Human gut faecalibacterium prausnitzii deploys a highly efficient conserved system to cross-feed on β-mannan-derived oligosaccharides. mBio. 2021;12:e0362820.
24. Culp EJ, Goodman AL. Cross-feeding in the gut microbiome: ecology and mechanisms. Cell Host Microbe. 2023;31:485-99.
25. Cartmell A, Muñoz-Muñoz J, Briggs JA, et al. A surface endogalactanase in Bacteroides thetaiotaomicron confers keystone status for arabinogalactan degradation. Nat Microbiol. 2018;3:1314-26.
26. Wang S, Mu L, Yu C, et al. Microbial collaborations and conflicts: unraveling interactions in the gut ecosystem. Gut Microbes. 2024;16:2296603.
27. Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490-5.
28. Shi E, Nie M, Wang X, et al. Polysaccharides affect the utilization of β-carotene through gut microbiota investigated by in vitro and in vivo experiments. Food Res Int. 2023;174:113592.
29. Holmes ZC, Villa MM, Durand HK, et al. Microbiota responses to different prebiotics are conserved within individuals and associated with habitual fiber intake. Microbiome. 2022;10:114.
30. Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560-4.
31. Wang Q, Spenkelink B, Boonpawa R, Rietjens IMCM, Beekmann K. Use of physiologically based kinetic modeling to predict rat gut microbial metabolism of the isoflavone daidzein to S-equol and its consequences for ERα activation. Mol Nutr Food Res. 2020;64:e1900912.
32. Bénard MV, Arretxe I, Wortelboer K, et al. Anaerobic feces processing for fecal microbiota transplantation improves viability of obligate anaerobes. Microorganisms. 2023;11:2238.
33. Salonen A, Nikkilä J, Jalanka-Tuovinen J, et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods. 2010;81:127-34.
34. Pjevac P, Hausmann B, Schwarz J, et al. An economical and flexible dual barcoding, two-step PCR approach for highly multiplexed amplicon sequencing. Front Microbiol. 2021;12:669776.
35. Berry D, Mader E, Lee TK, et al. Tracking heavy water (D2O) incorporation for identifying and sorting active microbial cells. Proc Natl Acad Sci U S A. 2015;112:E194-203.
36. Eichorst SA, Strasser F, Woyke T, Schintlmeister A, Wagner M, Woebken D. Advancements in the application of NanoSIMS and Raman microspectroscopy to investigate the activity of microbial cells in soils. FEMS Microbiol Ecol. 2015;91:fiv106.
37. Cui D, Kong L, Wang Y, Zhu Y, Zhang C.
38. Riva A, Rasoulimehrabani H, Cruz-Rubio JM, et al. Identification of inulin-responsive bacteria in the gut microbiota via multi-modal activity-based sorting. Nat Commun. 2023;14:8210.
39. Henry DG, Jarvis I, Gillmore G, Stephenson M, Emmings JF. Assessing low-maturity organic matter in shales using Raman spectroscopy: effects of sample preparation and operating procedure. Int J Coal Geol. 2018;191:135-51.
40. Caro TA, Kashyap S, Brown G, Chen C, Kopf SH, Templeton AS. Single-cell measurement of microbial growth rate with Raman microspectroscopy. FEMS Microbiol Ecol. 2024;100:fiae110.
41. Stöckel S, Kirchhoff J, Neugebauer U, Rösch P, Popp J. The application of Raman spectroscopy for the detection and identification of microorganisms. J Raman Spectrosc. 2016;47:89-109.
42. Lee KS, Pereira FC, Palatinszky M, et al. Optofluidic Raman-activated cell sorting for targeted genome retrieval or cultivation of microbial cells with specific functions. Nat Protoc. 2021;16:634-76.
43. Lee KS, Palatinszky M, Pereira FC, et al. An automated Raman-based platform for the sorting of live cells by functional properties. Nat Microbiol. 2019;4:1035-48.
44. Wright ES, Yilmaz LS, Noguera DR. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl Environ Microbiol. 2012;78:717-25.
45. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71-4.
46. R Studio Team. A language and environment for statistical computing. Available from: https://www.R-project.org. [Last accessed on 6 Aug 2025].
48. Oksanen J, Simpson GL, Blanchet FG, et al. Package ‘vegan’. Available from: https://cran.r-project.org/web/packages/vegan/vegan.pdf. [Last accessed on 6 Aug 2025].
49. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. 1995;57:289-300.
50. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
51. Letunic I, Bork P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024;52:W78-82.
52. Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327-36.
53. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. 2018;361:k2179.
54. O’Callaghan A, van Sinderen D. Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol. 2016;7:925.
55. Rinninella E, Raoul P, Cintoni M, et al. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms. 2019;7:14.
56. Hill C, Guarner F, Reid G, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11:506-14.
57. Hughes RL, Alvarado DA, Swanson KS, Holscher HD. The prebiotic potential of inulin-type fructans: a systematic review. Adv Nutr. 2022;13:492-529.
58. Scott KP, Gratz SW, Sheridan PO, Flint HJ, Duncan SH. The influence of diet on the gut microbiota. Pharmacol Res. 2013;69:52-60.
59. Bindels LB, Delzenne NM, Cani PD, Walter J. Towards a more comprehensive concept for prebiotics. Nat Rev Gastroenterol Hepatol. 2015;12:303-10.
60. Xiao M, Zhang C, Duan H, et al. Cross-feeding of bifidobacteria promotes intestinal homeostasis: a lifelong perspective on the host health. NPJ Biofilms Microbiomes. 2024;10:47.
61. González R, Klaassens ES, Malinen E, de Vos WM, Vaughan EE. Differential transcriptional response of Bifidobacterium longum to human milk, formula milk, and galactooligosaccharide. Appl Environ Microbiol. 2008;74:4686-94.
62. Liu M, Liu Z, Zhang N, et al. Preparation of polysaccharides from Crepis tectorum Linn. and the regulation effects on intestinal microbiota. Process Biochem. 2023;130:50-66.
63. Gao X, Xu F, Li T, et al. CAZymes-associated method to explore glycans that mitigate DSS-induced colitis via targeting Bacteroides cellulosilyticus. Int J Biol Macromol. 2024;258:128694.
64. Peterson CT, Sharma V, Iablokov SN, et al. 16S rRNA gene profiling and genome reconstruction reveal community metabolic interactions and prebiotic potential of medicinal herbs used in neurodegenerative disease and as nootropics. PLoS One. 2019;14:e0213869.
65. Aguirre M, Bussolo de Souza C, Venema K. The gut microbiota from lean and obese subjects contribute differently to the fermentation of arabinogalactan and inulin. PLoS One. 2016;11:e0159236.
66. He X, Zhao S, Li Y, Chen T.
67. Fujita K, Sasaki Y, Kitahara K. Degradation of plant arabinogalactan proteins by intestinal bacteria: characteristics and functions of the enzymes involved. Appl Microbiol Biotechnol. 2019;103:7451-7.
68. Hirmas B, Gasaly N, Orellana G, et al. Metabolic modeling and bidirectional culturing of two gut microbes reveal cross-feeding interactions and protective effects on intestinal cells. mSystems. 2022;7:e0064622.
69. Steinert RE, Rehman A, Sadabad MS, et al. Microbial micronutrient sharing, gut redox balance and keystone taxa as a basis for a new perspective to solutions targeting health from the gut. Gut Microbes. 2025;17:2477816.
70. Ku S, Haque MA, Jang MJ, et al. The role of Bifidobacterium in longevity and the future of probiotics. Food Sci Biotechnol. 2024;33:2097-110.
71. Wong CB, Odamaki T, Xiao J. Beneficial effects of Bifidobacterium longum subsp. longum BB536 on human health: modulation of gut microbiome as the principal action. J Funct Foods. 2019;54:506-19.
72. Gossling J, Moore WEC.
73. Salanitro JP, Muirhead PA, Goodman JR. Morphological and physiological characteristics of Gemmiger formicilis isolated from chicken ceca. Appl Environ Microbiol. 1976;32:623-32.
74. Zenner C, Hitch TCA, Riedel T, et al. Early-life immune system maturation in chickens using a synthetic community of cultured gut bacteria. mSystems. 2021;6:e01300-20.
75. Fitzgerald CB, Shkoporov AN, Sutton TDS, et al. Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genomics. 2018;19:931.
76. Guo W, Sun L, Yue H, et al. Associations of intermittent hypoxia burden with gut microbiota dysbiosis in adult patients with obstructive sleep apnea. Nat Sci Sleep. 2024;16:1483-95.
77. Qian L, Huang J, Qin H. Probiotics and dietary intervention modulate the colonic mucosa-associated microbiota in high-fat diet populations. Turk J Gastroenterol. 2020;31:295-304.
78. Arnold JW, Roach J, Fabela S, et al. The pleiotropic effects of prebiotic galacto-oligosaccharides on the aging gut. Microbiome. 2021;9:31.
79. Alberto F, Bignon C, Sulzenbacher G, Henrissat B, Czjzek M. The three-dimensional structure of invertase (beta-fructosidase) from Thermotoga maritima reveals a bimodular arrangement and an evolutionary relationship between retaining and inverting glycosidases. J Biol Chem. 2004;279:18903-10.
80. Andersen JM, Barrangou R, Abou Hachem M, et al. Transcriptional analysis of oligosaccharide utilization by Bifidobacterium lactis Bl-04. BMC Genomics. 2013;14:312.
81. Fujita K, Tsunomachi H, Lixia P, et al. Bifidobacterial GH146 β-L-arabinofuranosidase for the removal of β1,3-L-arabinofuranosides on plant glycans. Appl Microbiol Biotechnol. 2024;108:199.
82. Fujita K, Sakamoto A, Kaneko S, Kotake T, Tsumuraya Y, Kitahara K. Degradative enzymes for type II arabinogalactan side chains in Bifidobacterium longum subsp. longum. Appl Microbiol Biotechnol. 2019;103:1299-310.
83. Fujita K, Takashi Y, Obuchi E, Kitahara K, Suganuma T. Characterization of a novel β-L-arabinofuranosidase in Bifidobacterium longum: functional elucidation of a DUF1680 protein family member. J Biol Chem. 2014;289:5240-9.
84. Xu J, Xu R, Jia M, Su Y, Zhu W. Metatranscriptomic analysis of colonic microbiota’s functional response to different dietary fibers in growing pigs. Anim Microbiome. 2021;3:45.
85. Li W, Lin X, Liang H, et al. Genomic and functional diversity of the human-derived isolates of Faecalibacterium. Front Microbiol. 2024;15:1379500.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.