


Understanding cardiovascular aging as a disorder of mitochondrial network
 0
0 
Abstract
Cardiovascular aging is increasingly recognized as a mitochondrial-initiated systemic network dysfunction, a progressive, integrative failure driven by deteriorating mitochondrial quality and signaling. This review synthesizes emerging evidence linking comprehensive mitochondrial pathology to the erosion of cardiovascular resilience as a network-level dysfunction. Age-dependent remodeling of mitochondrial ultrastructure and component composition disrupts respiratory efficiency, positioning bioenergetic insufficiency as a central determinant of reduced stress tolerance across the cardiovascular system. Concurrently, defects in mitochondrial fission-fusion dynamics and impaired mitophagy propagate dysfunction within the mitochondrial network, amplifying
Keywords
INTRODUCTION
Aging remains the dominant risk factor for cardiovascular disease[1,2], yet energetic biology is still too often treated as secondary to structural pathology. Here, we argue that cardiovascular aging is fundamentally a disorder of the mitochondrial network, characterized by declining bioenergetic reserve, impaired quality control, and maladaptive organelle-to-nucleus and organelle-to-immune signaling that progressively reduces cardiac and vascular resilience[2-5]. The implication is practical: delaying disease onset and preserving function will require preserving mitochondrial performance under stress, including efficient flux capacity, effective turnover, and controlled stress signaling.
The heart is uniquely susceptible to mitochondrial network disorders because mitochondria constitute a substantial proportion of cardiomyocyte volume and operate as an integrated cellular network that must synchronously sustain adenosine triphosphate (ATP) production, calcium handling, redox balance, and survival signaling on a beat-to-beat basis[3,6,7]. In aged rodent hearts and human myocardial samples, coordinated changes in cristae architecture and cardiolipin composition impair electron transport efficiency[3,8] and energetic coupling[8,9], while dysregulation of mitochondrial fission-fusion dynamics and insufficient mitophagy disrupt network quality control, allowing dysfunctional mitochondria to persist and propagate failure signals[10,11]. Importantly, these abnormalities extend beyond bioenergetics[6,12]. In murine models of cardiac injury and in cultured cardiomyocytes, damaged mitochondria release nucleic acids and peptides that activate innate immune pathways such as cyclic guanosine monophosphate-adenosine monophosphate (GMP-AMP) synthase-stimulator of interferon genes (cGAS-STING)[13], linking mitochondrial dysfunction to chronic inflammation and impaired stress adaptation across cardiomyocytes, endothelial cells, and fibroblasts[12,13]. Together, these observations support the concept that cardiac aging and disease reflect not isolated mitochondrial defects, but a breakdown of mitochondrial-cellular network communication that progressively destabilizes tissue-level homeostasis.
An important conceptual distinction is whether mitochondrial dysfunction is a primary driver of cardiovascular aging or a downstream consequence of cumulative cellular damage. Current evidence supports a bidirectional model. In experimental systems, genetically or pharmacologically induced mitochondrial defects are sufficient to recapitulate key aging phenotypes, including reduced stress tolerance, impaired diastolic relaxation, endothelial dysfunction, and heightened inflammatory signaling, indicating that mitochondrial dysfunction can act as an initiating event[14,15]. Conversely, canonical aging processes such as telomere attrition, genomic instability, proteostatic decline, and chronic inflammatory stress progressively impair mitochondrial quality control, bioenergetics, and redox balance[14,16,17]. In this context, mitochondrial dysfunction becomes both a cause and an amplifier of aging phenotypes. The cardiovascular system is particularly susceptible to this feed-forward architecture because high energetic demand and continuous mechanical stress render mitochondria early sensors of strain and late executors of maladaptive remodeling. Thus, rather than framing mitochondrial decline strictly as cause or consequence, we interpret it as a rate-limiting node within a self-reinforcing aging network.
Clinically, the signature of this network failure is diffuse rather than focal: diastolic dysfunction precedes systolic loss[18], microvascular rarefaction and endothelial stiffness impair perfusion, atrial substrate ages into fibrillation, and stress intolerance emerges years before overt heart failure. Across models and human studies, impaired bioenergetic reserve rather than resting ATP alone tracks most closely with functional decline. This shifts therapeutic priorities: the target is not “less reactive oxygen species (ROS)”, but more flexible, high-quality mitochondria capable of rapid flux, effective turnover, and calibrated retrograde signaling.
Promising avenues span behavior, pharmacology, and emerging biologics. Exercise and nutritional strategies can enhance mitochondrial biogenesis and turnover[3,19,20]. Agents that boost nicotinamide adenine dinucleotide (NAD+) or activate adenosine monophosphate -activated protein kinase (AMPK)/sirtuins (SIRTs) may restore the redox/acetylation balance[21,22]. Mitophagy inducers and cardiolipin-stabilizing approaches aim to improve organelle quality[23]. Endothelial-targeted interventions seek to rescue the microcirculation that governs myocardial oxygen delivery[12]. Yet translation has lagged because trials seldom measure mitochondrial function directly, interventions often ignore cell-type and subcellular heterogeneity (intermyofibrillar vs. subsarcolemmal pools; cardiomyocyte vs. endothelial mitochondria), and endpoints capture downstream pathology rather than mitochondrial youthfulness.
This review synthesizes a network-centric view of cardiovascular aging and organizes the field around three axes: energy, quality control, and signaling to unify disparate observations. We propose practical metrics to quantify “mitochondrial age”, highlight crosstalk between cardiovascular mitochondria and remote organs as an early and tractable target, and outline a framework for cellular mitochondrial crosstalk and its translational implications that prioritizes function over surrogate biomarkers. Making the heart’s mitochondria functionally younger, rather than merely slowing the organismal aging, should become a central objective of cardiovascular aging research and therapy.
KEY RECOGNITIONS IN MITOCHONDRIAL NETWORKS AND CARDIAC AGING
Mitochondrial networks as an organizing principle
An important recognition emerging from the past decade is that mitochondrial dysfunction in the aging heart is not well captured by single readouts of “mitochondrial content”, resting ATP levels, or bulk oxidative stress, but instead reflects failure modes of a spatially organized mitochondrial network[24,25]. Cardiac mitochondria operate as an interconnected and dynamically reconfiguring system that must match energy production to rapidly fluctuating demands while simultaneously coordinating calcium handling, redox buffering, and stress signaling. Aging perturbs these network properties through cumulative alterations in ultrastructure, membrane composition, and protein homeostasis[26] with downstream consequences that depend on network topology and connectivity rather than on the mean function alone[24,27,28]. This perspective provides a mechanistic basis for why bioenergetic reserve and stress responsiveness can decline despite apparently preserved baseline energetics, and it reframes mitochondrial pathology as a systems-level determinant of reduced myocardial resilience rather than as an epiphenomenon of end-stage disease.
Heterogeneity, crosstalk, and network failure across cardiac cell types
A second key recognition is that mitochondrial network dysfunction in cardiac aging is intrinsically heterogeneous, both among mitochondrial subpopulations and within the cellular myocardial ecosystem[3,29]. Intermyofibrillar and subsarcolemmal mitochondrial pools exhibit distinct biophysical constraints, substrate access, and turnover dynamics, and these differences likely shape how energetic insufficiency and quality-control failure manifest during aging[30,31]. Moreover, cardiomyocyte mitochondrial decline interacts with, and may be initiated or amplified by, parallel dysfunction in endothelial and microvascular mitochondria that govern perfusion-metabolism coupling and nitric oxide (NO) signaling, thereby influencing early diastolic impairment and ischemic susceptibility[12,32,33]. Aging-associated mitochondrial stress also propagates through retrograde communication pathways, including mitochondrial-derived ligands that activate innate immune sensing and remodel transcriptional programs, linking organelle dysfunction to inflammaging, fibrosis, and maladaptive remodeling[13,34]. Together, these insights position the mitochondrial network distributed across cell types and integrated through metabolic and inflammatory crosstalk as a central explanatory framework for cardiac aging and a rational substrate for interventions aimed at preserving function.
In addition to direct cellular interactions, recent studies have recognized the role of mitochondrial transfer and extracellular vesicles (EVs) as significant mechanisms of intercellular mitochondrial communication in cardiac aging[35-37]. Intercellular mitochondrial transfer refers to the process through which functional mitochondria or mitochondrial components are transferred from donor cells to recipient cells, allowing for mitochondrial rescue or reprogramming. This form of mitochondrial transplantation occurs through direct cell-to-cell connections (such as gap junctions) or via vesicular transfer, with emerging evidence suggesting that damaged or stressed cells can export mitochondria to healthy cells to mitigate dysfunction[38,39]. Mitochondria are also transported in EVs, including both small EVs (such as exosomes) and larger vesicles, known as exospheres, which are capable of packaging entire mitochondria or mitochondrial components[35,40,41]. These vesicles serve as carriers of mitochondrial material and signaling molecules, facilitating the transfer of mitochondria between cells and enabling the adaptation of recipient cells to metabolic stress[42,43]. This process not only promotes mitochondrial rejuvenation but also helps coordinate cellular responses to stress, inflammation, and metabolic demands[36,37,43]. Recent findings suggest that the transfer of mitochondria via EVs can influence cardiomyocyte function, endothelial cell behavior, and even the microvascular environment, providing a novel layer of complexity in the understanding of intercellular communication and network dysfunction in cardiac aging[39,43]. The ability of mitochondria to be transferred via these mechanisms represents a form of mitochondrial network maintenance, and dysfunction in this process may contribute to the maladaptive remodeling observed in aging and heart disease.
CURRENT VIEW: CARDIOVASCULAR AGING AND MITOCHONDRIAL IMBALANCE
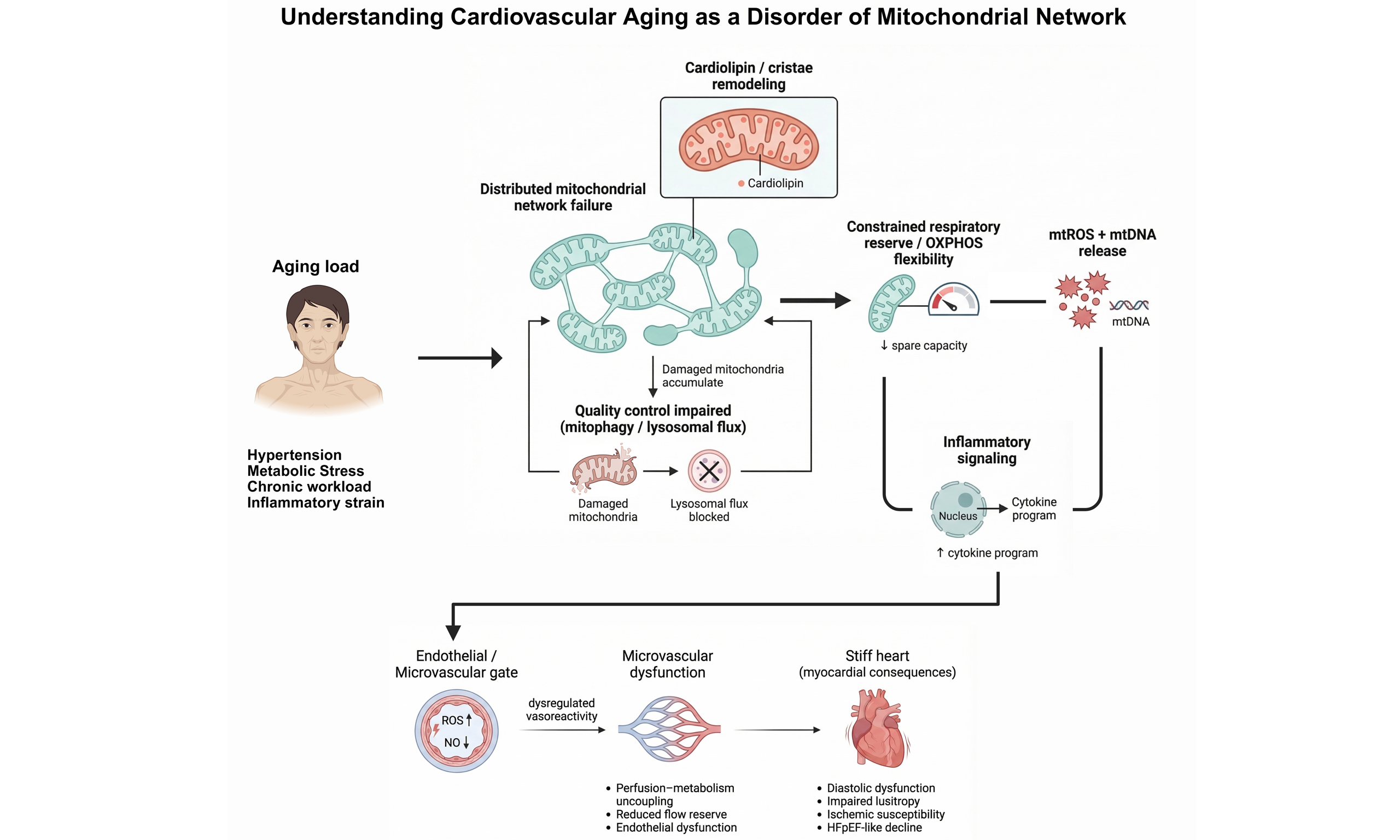
A prevailing framework now interprets cardiovascular aging as a progressive mitochondrial imbalance in which the capacity to generate ATP adaptively, maintain organelle integrity, and constrain stress-evoked signaling becomes insufficient to meet the cumulative workload and damage[44]. This view is supported by the observation that aging phenotypes are expressed most clearly as deficits in functional reserve, exercise intolerance, reduced ischemic tolerance, and endothelial dysfunction rather than as isolated reductions in resting ATP[45]. Mechanistically, mitochondrial imbalance is increasingly defined by three interlocking features: (i) constrained oxidative phosphorylation response to demand[3], (ii) reduced renewal throughput (dynamics and mitophagy)[46], and (iii) amplified retrograde signaling that biases tissues toward sterile inflammation and remodeling[5,34,47] [Figure 1]. Importantly, this model accommodates the multi-organelle nature of aging while proposing that mitochondria function as a rate-limiting integrator of energetic, redox, and inflammatory cues across cardiomyocytes and the vascular wall.
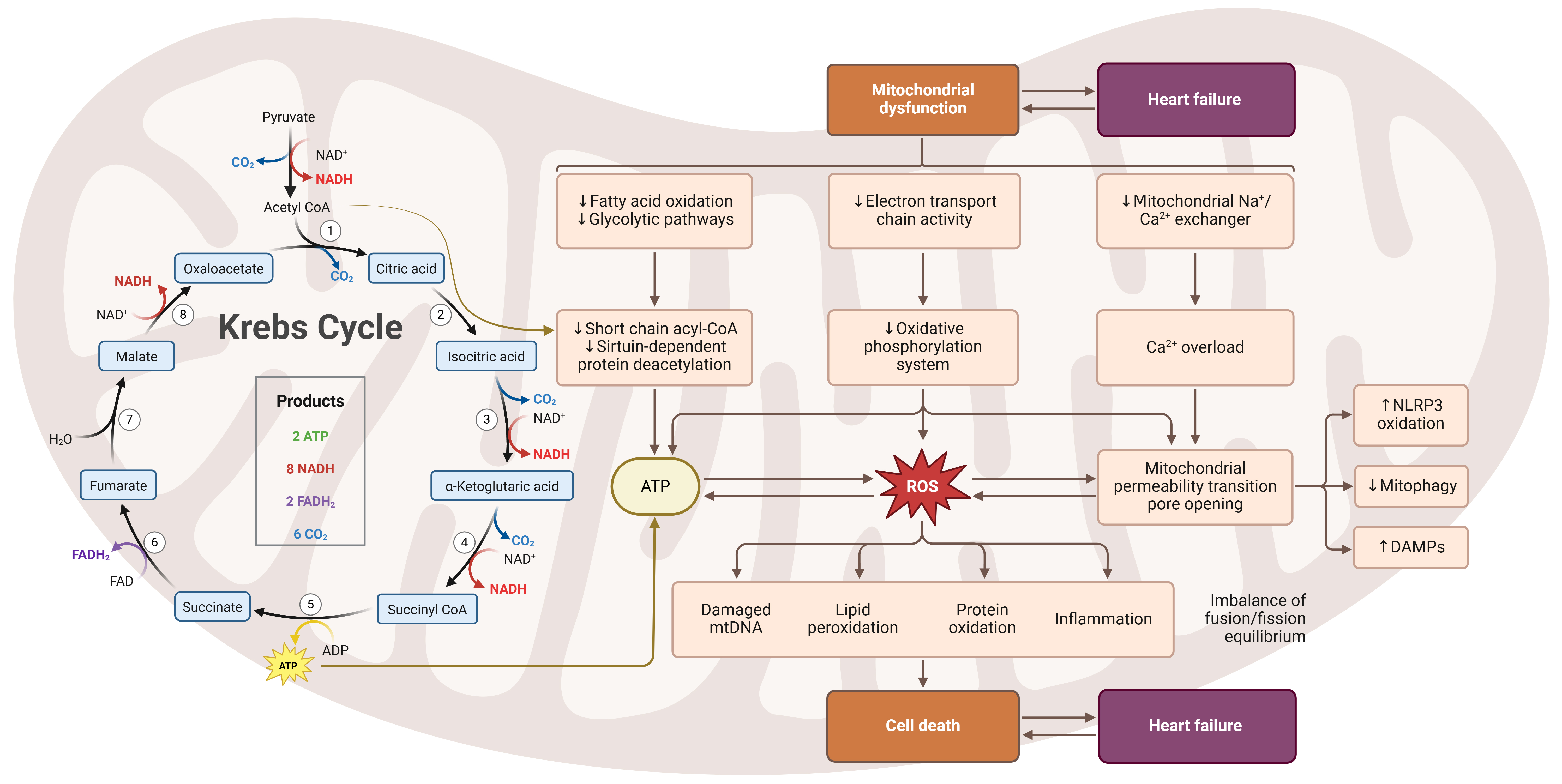
Figure 1. Mitochondrial metabolic dysfunction and redox-driven injury pathways in cardiovascular aging resulting in heart failure. (Created in BioRender. Ahmad D (2026) https://BioRender.com/61c7ow3). NADH: Nicotinamide adenine dinucleotide; FAD: flavin adenine dinucleotide; ATP: adenosine triphosphate; mtDNA: mitochondrial DNA; DAMPs: danger-associated molecular patterns; ROS: reactive oxygen species; NLRP3: nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3
Energetic constraint: inner membrane remodeling and metabolic control
In aged rodent myocardium and ex vivo human cardiac tissue studies, age-related energetic decline is increasingly attributed to mechanisms that restrict peak respiratory flux and coupling efficiency[3]. Structural remodeling of the inner mitochondrial membrane (IMM), including disrupted cristae architecture and altered membrane composition, can impair electron transport organization and elevate electron leak under stress[48]. Cardiolipin is a key molecular node because it stabilizes multiple respiratory complexes and supports inner-membrane curvature; remodeling or oxidation of cardiolipin can therefore reduce oxidative phosphorylation efficiency and contribute to decreased stress tolerance[49]. In parallel, aging perturbs metabolic control systems that tune mitochondrial enzymes and redox state[50]. Declining NAD+ availability and altered downstream signaling [SIRT-dependent deacetylation programs, adenosine monophosphate-activated protein kinase (AMPK)-linked nutrient sensing] alter mitochondrial protein acetylation, substrate oxidation, and antioxidant defense capacity, thereby narrowing the dynamic range of mitochondrial output[50,51]. The net effect is a disproportionate loss of reserve capacity that is most evident during physiological challenge rather than at baseline.
Turnover bottlenecks: dynamics, mitophagy, and lysosomal throughput
Mitochondrial quality control depends on coordinated processes that preserve organelle structure, segregate damaged components, and remove dysfunctional mitochondria. Mitochondrial dynamics refers to the continuous cycles of fission and fusion that remodel the mitochondrial network, allowing content mixing, dilution of damage, and isolation of dysfunctional segments. These processes are governed primarily by large GTPases, including dynamin-related protein 1 (DRP1) (fission), mitofusin 1/2 (MFN1/2), and optic atrophy 1 (OPA1) (fusion), and are essential for maintaining bioenergetic efficiency and structural integrity in highly energy-demanding tissues such as the heart[52-54]. Mitophagy complements these processes by selectively removing damaged or bioenergetically compromised mitochondria through phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)/Parkin-dependent ubiquitin signaling or receptor-mediated pathways such as BCL2 interacting protein 3 (BNIP3)/NIP3-like protein X (NIX) and FUN14 domain-containing 1 (FUNDC1). Together, mitochondrial dynamics and mitophagy form an integrated quality-control system that preserves respiratory reserve, limits excessive ROS production, and restrains inflammatory signaling. Disruption of either arm has been strongly implicated in cardiovascular aging and disease[54-57].
A second core association is the reduction in effective mitochondrial renewal relative to damage burden. Mitochondrial fission-fusion machinery, including DRP1, MFN1/2, and OPA1, maintains network function by enabling content complementation, segregation of damaged segments, and coordination with biogenesis and autophagic clearance[58]. Aging frequently perturbs this coupling, promoting fragmentation and impairing functional complementation, while mitophagy flux becomes insufficient to remove dysfunctional or damaged organelles[46,59]. This insufficiency can arise at several points, including mitochondrial damage sensing and ubiquitin signaling (PINK1/Parkin)[60], receptor-mediated pathways such as BNIP3/NIX, FUNDC1[61], or downstream autophagosome-lysosome clearance, yet it converges on persistence of dysfunctional mitochondria that depress respiratory reserve and amplify redox stress[62,63]. This perspective is particularly important in aging because it suggests that interventions must restore quality-control flux rather than merely increase mitochondrial content.
Integration of mitochondrial quality control systems
Mitochondrial quality control in the aging cardiovascular system is best understood not as a collection of independent pathways but as an integrated network of interdependent regulatory modules. Core components of this network include mitochondrial dynamics (the balance between fission and fusion), mitophagy, metabolic signaling pathways such as NAD+-dependent SIRT signaling and AMPK signaling, and lysosomal clearance capacity. These systems are tightly coupled such that dysfunction in one node can propagate failure across the network[52]. For example, declining NAD+ availability with aging impairs the activity of SIRT deacetylases, particularly SIRT1 and SIRT3, which regulate transcriptional programs controlling mitochondrial biogenesis and autophagic turnover. Reduced SIRT signaling therefore diminishes the transcriptional and post-translational regulation of mitophagy components, including PINK1/Parkin and receptor-mediated pathways such as BNIP3 and FUNDC1[51,64]. In parallel, energetic stress and impaired AMPK activation can reduce mitochondrial fission events necessary for isolating damaged mitochondrial segments prior to autophagic removal[65,66]. When these regulatory layers fail simultaneously, dysfunctional mitochondria accumulate, producing excess ROS and releasing mitochondrial danger signals that further amplify inflammatory and metabolic stress. This interdependence suggests that mitochondrial aging is not simply an additive decline of individual pathways but rather a systems-level collapse of quality-control coordination. Consequently, therapeutic strategies that restore upstream regulators of mitochondrial maintenance, such as NAD+ metabolism or AMPK signaling, may have disproportionately broad effects by simultaneously improving mitochondrial turnover, metabolic flexibility, and inflammatory control [Table 1].
Integration of mitochondrial quality control pathways in cardiovascular aging
| System | Key components | Function | Failure mechanism in aging | Downstream consequence |
| NAD+/Sirtuin signaling | NAD+, SIRT1, SIRT3 | Regulates mitochondrial metabolism, antioxidant defense, and transcription of mitochondrial genes | Age-dependent NAD+ decline reduces sirtuin activity | Impaired mitochondrial biogenesis and reduced mitophagy signaling |
| AMPK energy sensing | AMPK, PGC-1α | Detects energetic stress and stimulates mitochondrial biogenesis and autophagy | Reduced AMPK responsiveness in aging | Decreased mitochondrial turnover and metabolic flexibility |
| Mitochondrial dynamics | DRP1, MFN1/2, OPA1 | Controls mitochondrial fission and fusion to segregate damaged organelles | Dysregulated dynamics promote fragmentation or hyperfusion | Impaired isolation of damaged mitochondria |
| Mitophagy pathways | PINK1/Parkin, BNIP3/NIX, FUNDC1 | Selective removal of dysfunctional mitochondria | Reduced mitophagy signaling and lysosomal flux | Accumulation of dysfunctional mitochondria |
| Lysosomal degradation | Autophagosome–lysosome pathway | Final degradation step for mitochondrial clearance | Lysosomal decline with aging | Incomplete mitochondrial turnover and inflammatory signaling |
Mitochondrial communication networks and vascular gating
Mitochondrial communication in cardiovascular aging is enabled by the organelle’s compartmental design and by specialized interfaces that couple bioenergetic state to cytosolic, nuclear, and extracellular programs. Structurally, the outer mitochondrial membrane (OMM) defines a signaling and trafficking boundary enriched in permeability and docking proteins, the intermembrane space (IMS) functions as a redox-sensitive relay compartment, and the IMM is folded into cristae that spatially organize electron transport, proton pumping, and ATP synthase into high-efficiency microdomains[67]. The matrix contains the tricarboxylic acid (TCA) cycle, pathways for handling fatty-acid-derived acetyl units, one-carbon metabolism modules, and the mitochondrial genome packaged into nucleoids[68]. This architecture matters because most “messages” originate as compartment-specific deviations in membrane potential, reduced nicotinamide adenine dinucleotide (NADH)/NAD+ balance, ROS tone, calcium (Ca2+) flux, and metabolite pools. These changes are generated locally and are exported or transduced across membranes through defined conduits, rather than by diffuse equilibration[68].
At the sub-mitochondrial level, metabolic crosstalk is dominated by a matrix-to-IMM coupling loop in which energetic demand, redox tone, and Ca2+ handling are integrated across sub-mitochondrial domains [Figure 2]. In this loop, matrix dehydrogenase activity sets reducing-equivalent supply, while IMM respiratory flux sets demand and leak[69]. Ca2+ uptake is a key control knob for this loop because mitochondrial Ca2+ entry tunes Ca2+-sensitive matrix dehydrogenases and thereby adjusts NADH regeneration during stress[70], while dysregulated Ca2+ handling increases ROS production and destabilizes energetic control[70], shifting mitochondria from adaptive signaling to damage amplification. The relevance for vascular aging is that endothelial mitochondria often operate near signaling thresholds, where modest shifts in redox or Ca2+ microdomains can reprogram NO bioavailability, inflammatory tone, and barrier behavior[12].
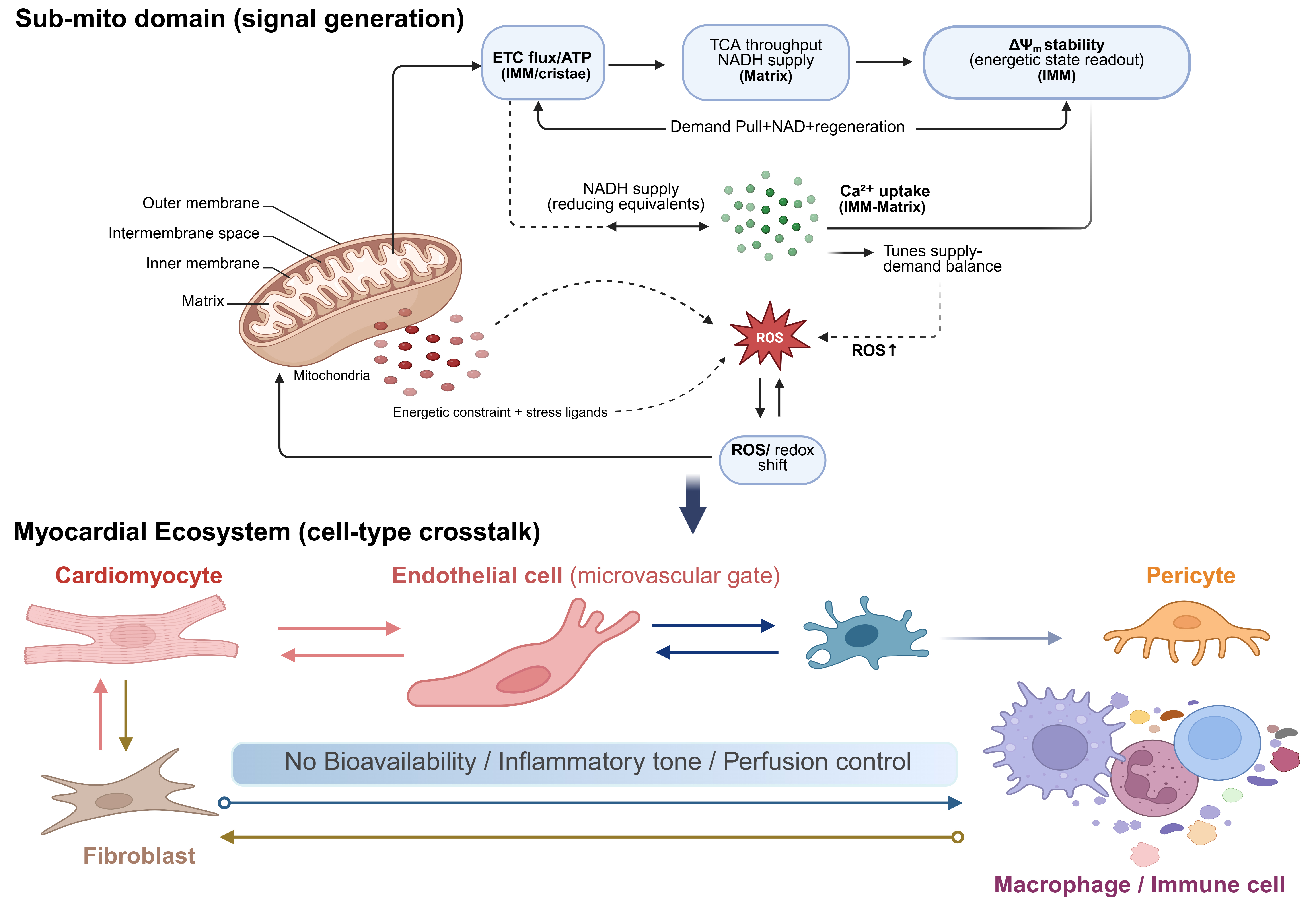
Figure 2. Sub-mitochondrial signal generation and myocardial cell-type crosstalk in cardiovascular aging. (Created in BioRender. Shila TA (2026) https://BioRender.com/2bztp85). ETC: Electron transport chain; IMM: inner mitochondrial membrane; ATP: adenosine triphosphate; TCA: tricarboxylic acid; NADH: nicotinamide adenine dinucleotide; ROS: reactive oxygen species.
High-bandwidth organelle-to-organelle crosstalk is then implemented through physical contact sites rather than through bulk diffusion[71] [Figure 2]. The best-characterized interface is the endoplasmic reticulum (ER)-mitochondria contact domain, often termed the mitochondria-associated membrane (MAM)[71], where ER Ca2+ release channels are functionally coupled to OMM uptake routes[72]. A canonical example is the inositol 1,4,5-trisphosphate receptor (IP3R)-glucose-regulated protein 75 (GRP75)-voltage-dependent anion channel 1 (VDAC1) axis, which positions ER Ca2+ release near mitochondrial entry pathways so that mitochondria can sample high local Ca2+ microdomains despite the low affinity of downstream uptake machinery[72]. This arrangement supports physiological metabolic matching, but it also creates a direct route through which chronic ER stress or dysregulated Ca2+ release can drive mitochondrial Ca2+ overload, ROS escalation, and downstream inflammatory signaling[72].
Quality control and cargo routing provide a second inter-organelle communication layer that is essential for interpreting which mitochondrial components are presented to the cell. When damage is focal or sublethal, mitochondria can generate mitochondria-derived vesicles (MDVs) that selectively package oxidized proteins or other cargo for trafficking to endolysosomal compartments[73]. This process provides a rapid, graded disposal pathway that can precede or complement whole-organelle mitophagy. Importantly, MDV biology also links mitochondrial quality control to immune signaling and to EV composition, indicating that mitochondrial communication includes selective export decisions, not only internal degradation choices[74].
Retrograde signaling to the nucleus can be conceptualized as three partially separable output classes. The first is metabolite-to-epigenome coupling. Mitochondrial metabolism supplies or regulates cofactors and intermediates that directly constrain chromatin-modifying enzymes and transcriptional competence, including acetyl-Co-enzyme A, α-ketoglutarate, succinate, fumarate, and NAD+-linked pathways. In practical terms, mitochondrial flux states can bias the capacity for histone acetylation and demethylation, thereby shifting transcriptional set points even in the absence of overt cell death or inflammation[75]. This coupling provides a mechanistic bridge from mitochondrial substrate handling to durable vascular and myocardial remodeling programs. The second is stress transduction via dedicated relay pathways, exemplified by DAP3 binding cell death enhancer 1 (DELE1)-dependent activation of the heme-regulated inhibitor (HRI) branch of the integrated stress response during mitochondrial import or proteostasis perturbations[76]. This pathway converts mitochondrial distress into coordinated translational and transcriptional remodeling, including endocrine-like outputs, and helps explain how chronic, sublethal mitochondrial stress can become a stable cellular phenotype[76]. The third output class is innate immune activation when compartmental containment fails. In murine models of pressure overload, myocardial infarction, and in vitro cardiomyocyte injury, the release of oxidized mitochondrial DNA (mtDNA) into the cytosol can activate cGAS-STING signaling and downstream interferon and nuclear factor kappa B (NF-κB) programs, linking mitochondrial injury to sterile inflammation, fibroblast activation, and extracellular matrix remodeling in cardiovascular disease settings[77].
These mechanistic layers also clarify why intact mitochondria generally do not circulate in blood as stable, freely functional units. Outside the cellular environment, mitochondria are deprived of the ionic buffering, substrate provisioning, and chaperone-supported membrane maintenance required for sustained oxidative phosphorylation[78]. In addition, mitochondrial surfaces and contents are immunologically conspicuous because mitochondrial molecules share features with bacterial motifs, making extracellular mitochondria prone to being interpreted as danger signals and rapidly cleared[79]. Consistent with this, bioenergetic characterization of circulating cell-free mitochondria in human blood argues against robust respiratory competence in vivo, supporting a model in which most extracellular mitochondrial material reflects release and processing rather than the purposeful circulation of fully functional organelles.
What circulates effectively are mitochondrial signals in forms that are smaller, stabilized, or packaged[36]. These include mtDNA fragments, mitochondrial EVs, and stress-induced mitokines that propagate cardiac mitochondrial stress to distal organs [Figure 3]. One major class is cell-free mitochondrial nucleic acids, particularly mtDNA fragments, which can act as inflammatory ligands and have emerged as biomarkers of tissue stress and immune activation in blood[79]. The second class is vesicle-associated mitochondrial cargo. Reviews describe mitochondrial extracellular vesicles (mitoEVs) and related vesicular populations as mediators of immune responses and bioenergetic remodeling, with growing emphasis on their potential as aging biomarkers[36]. Mechanistic work further supports that selective packaging of mitochondrial proteins into EVs can depend on MDV pathways, implying that mitochondrial material can be exported in a regulated, pathway-specific manner rather than by nonspecific rupture alone[74]. A third class comprises endocrine-like “mitokines” induced by mitochondrial stress programs, most prominently fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15), which are increasingly positioned as systemic readouts of mitochondrial stress and as mediators of organism-level metabolic adaptation but may become maladaptive when chronically elevated[80].
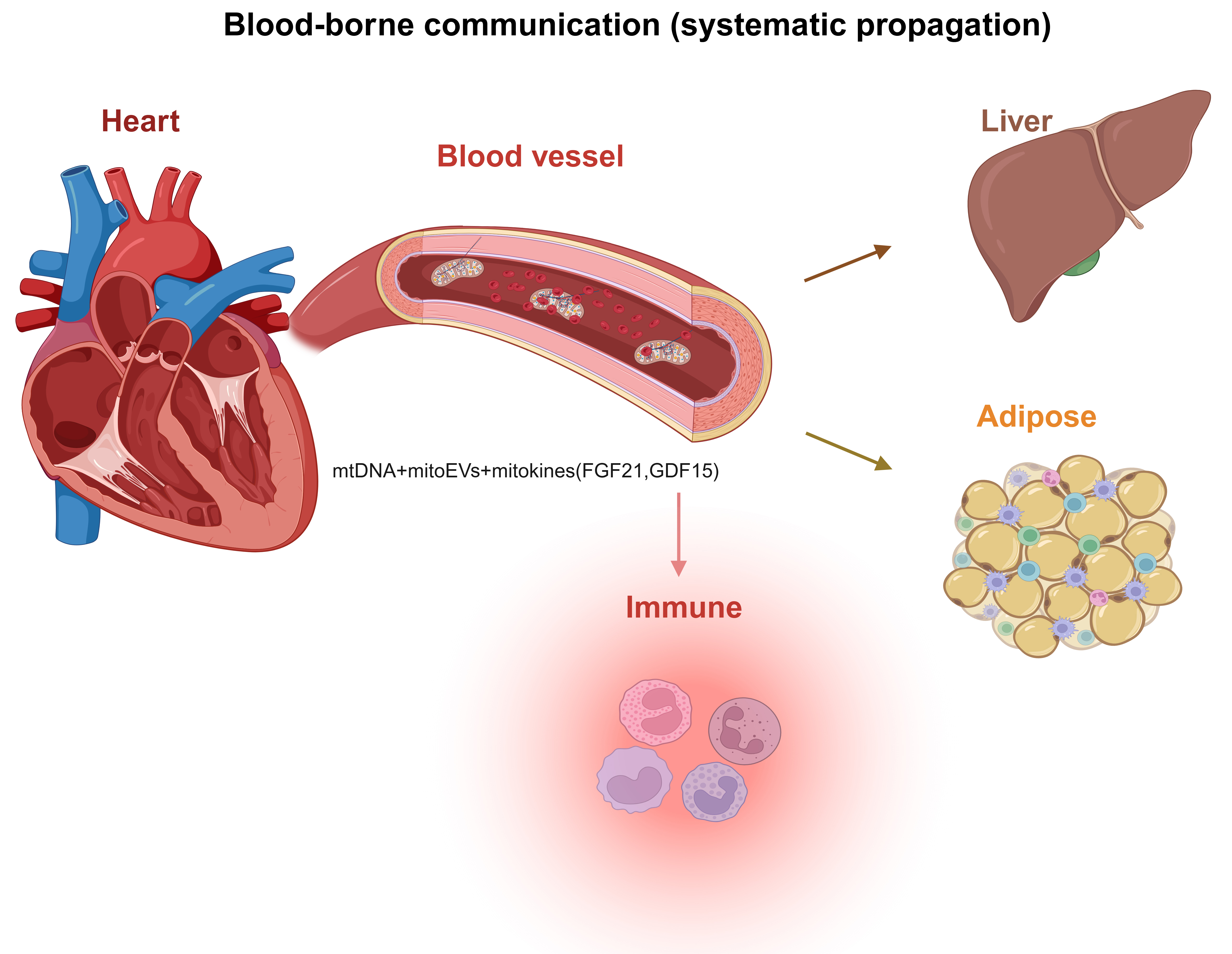
Figure 3. Blood-borne propagation of mitochondrial stress signals from the heart to peripheral organs. (Created in BioRender. Shila TA (2026) https://BioRender.com/94twtl3). mitoEVs: Mitochondrial extracellular vesicles; mtDNA: mitochondrial DNA; FGF21: fibroblast growth factor 21; GDF15: growth differentiation factor 15.
In this framework, the mitochondrial network communicates with multiple partners across scales, and vascular gating arises because endothelial and microvascular cells sit at a convergence point for metabolic sensing, inflammatory routing, and flow control. Intracellularly, endothelial mitochondria integrate ER-derived Ca2+ microdomains at MAMs with redox control and quality-control routing, shaping NO biology and inflammatory responsiveness. Tissue-level propagation occurs through paracrine cytokines, vesicle traffic, and immune recruitment[81] but also through direct microvascular cell-to-cell conduits that can transmit metabolic influence. For example, pericyte-to[82]-endothelial communication via tunneling nanotubes has been described with measurable effects on endothelial metabolism[83], emphasizing that microvascular energetic phenotypes can be coordinated through physical intercellular structures rather than through soluble mediators alone. Systemically, blood-borne mtDNA, mitoEV cargo, and mitokines couple local mitochondrial stress to distal immune and metabolic responses, creating feedback loops that can stabilize sterile inflammation and impair perfusion-energy matching when mitochondrial injury becomes persistent.
Mitochondria as central drivers of cardiovascular aging
A useful conceptual distinction is that mitochondrial signaling during aging evolves from an initially adaptive stress-response program to a maladaptive amplifier of tissue dysfunction. In the early stages of cardiovascular aging, moderate mitochondrial stress can activate protective signaling pathways, including transient increases in ROS that stimulate antioxidant defenses, enhanced mitochondrial turnover through mitophagy, and induction of mitochondrial stress responses such as mitokine signaling (e.g., FGF21 and GDF15). These responses can temporarily preserve cellular homeostasis by promoting metabolic flexibility and the removal of damaged organelles[84,85]. However, as aging progresses and mitochondrial damage accumulates, these same pathways become chronically activated. Persistent mtDNA release, sustained cGAS-STING signaling, and impaired mitophagy shift mitochondrial signaling toward a maladaptive state characterized by sterile inflammation, endothelial dysfunction, and reduced bioenergetic reserve. In this later stage, mitochondrial signaling no longer supports adaptation but instead reinforces fibrotic remodeling, microvascular dysfunction, and impaired cardiac stress tolerance[5,86].
Mitochondrial network dysfunction also interacts closely with traditional cardiovascular risk factors, particularly hypertension and diabetes. Chronic pressure overload in hypertension increases myocardial energetic demand while simultaneously promoting mitochondrial oxidative stress and endothelial dysfunction, which together impair NO signaling and vascular relaxation[87,88]. Similarly, in diabetes and insulin resistance, mitochondrial substrate overload and altered fatty acid oxidation drive excessive electron transport chain (ETC) flux and ROS generation, contributing to mtDNA damage and impaired quality-control pathways. These cardiometabolic conditions, therefore, accelerate mitochondrial aging by compounding defects in redox regulation, NAD+ signaling, and mitochondrial turnover, ultimately reducing the capacity of the mitochondrial network to maintain perfusion-energy coupling under physiological stress[89].
A growing body of evidence places mitochondria at the center of cardiovascular aging. In the heart and vasculature, aging is accompanied by declining respiratory efficiency[3], remodeling of cristae and cardiolipin[3], imbalance in fission-fusion dynamics, and shortfalls in mitophagy[46]. These changes erode bioenergetic reserve and blunt stress-adaptative responses, which are more accurately reflected by dynamic mitochondrial capacity than by resting ATP levels alone. Recent syntheses frame these alterations as a coordinated network rather than isolated defects[56], shifting the field’s focus beyond ROS toward mitochondrial quality control and flexibility. Vascular aging provides an early, clinically salient readout of this biology. Large-artery stiffening and endothelial dysfunction emerge through intertwined pathways linking inflammation, redox imbalance, and dysregulated energy-sensing nodes (e.g., AMPK and SIRTs). At the microvascular level, rarefaction and endothelial senescence impair perfusion-metabolism matching and are increasingly recognized in phenotypes such as heart failure with preserved ejection fraction (HFpEF).
Mitochondria also act as immunometabolic signaling hubs. Leakage of mtDNA and other danger signals can activate cGAS-STING and related pathways, sustaining the sterile inflammation typical of aging tissues, including heart and vessels[5]. This signaling axis is currently the main focus of therapeutic exploration in cardiovascular disease. The current review synthesizes evidence across epidemiology, clinical research, and experimental cardiology to (i) frame cardiovascular aging as a disorder of an integrated mitochondrial vascular network, (ii) distill advances in mitochondrial quality control, bioenergetic reserve, and innate immune signaling as they pertain to cardiac and vascular aging, and (iii) outline implications for prevention and therapy in older adults, with particular attention to strategies that enhance mitochondrial flexibility and microvascular function. By integrating these strands, this review seeks to identify practical opportunities for measurement and intervention that can improve cardiovascular health span.
Therapeutic opportunities for mitochondrial aging in cardiovascular disease
Therapeutic strategies for mitochondrial aging in cardiovascular disease are shifting from broad, nonspecific “mitochondrial support” toward interventions that target defined mitochondrial pathways and can be evaluated with measurable human endpoints. In vascular aging, mitochondria-targeted redox modulation has advanced beyond preclinical rationale: chronic supplementation with the mitochondrial antioxidant mitoquinone (MitoQ) improved brachial artery flow-mediated dilation and reduced aortic stiffness in healthy late middle-aged and older adults in a randomized controlled study, and this line of work has expanded into protocols designed to test vascular function outcomes in older cohorts.
In parallel, quality-control therapeutics that enhance mitochondrial turnover have entered human testing. Urolithin A, a mitophagy-linked intervention, improved muscle endurance and shifted circulating biomarkers consistent with enhanced mitochondrial health in randomized trials in older adults, offering proof-of-concept that organelle quality control can be modulated safely with functional readouts in humans[90]. NAD+-repletion strategies similarly demonstrate reliable target engagement in humans, and recent translational efforts increasingly evaluate blood pressure and arterial stiffness as mechanistically relevant vascular surrogates; a pilot randomized clinical trial combining nicotinamide riboside (NR) with supervised exercise in middle-aged and older adults with hypertension exemplifies this mechanistic, endpoint-linked approach[91].
Dietary timing interventions are also being tested with mechanistic vascular endpoints in older populations. Notably, months-long time-restricted eating protocols have been designed specifically to examine whether sustained adherence improves endothelial function and neurovascular or cerebrovascular coupling in community-dwelling older adults[92].
At the pharmacologic end of the spectrum, inner-membrane and cristae-directed stabilization reached a major regulatory milestone in 2025, when the U.S. Food and Drug Administration granted accelerated approval to Forzinity (elamipretide) injection for Barth syndrome, highlighting the clinical tractability of therapies that directly target mitochondrial inner-membrane structure and function[93]. Finally, established cardiometabolic agents are increasingly interpreted through mitochondrial and endothelial mechanisms, with evidence that sodium-glucose cotransporter 2 (SGLT2) inhibition can improve endothelial cell bioenergetics and reduce mitochondrial oxidative stress in mechanistic studies. This supports a pragmatic pathway in which clinically validated drugs may deliver partial “mitochondrial rejuvenation” alongside improvements in clinical outcomes[94]. Collectively, these approaches align with the mechanistic therapeutic objectives illustrated in Figure 4, targeting mitochondrial structure, reserve capacity, metabolic coupling, and stress tolerance.
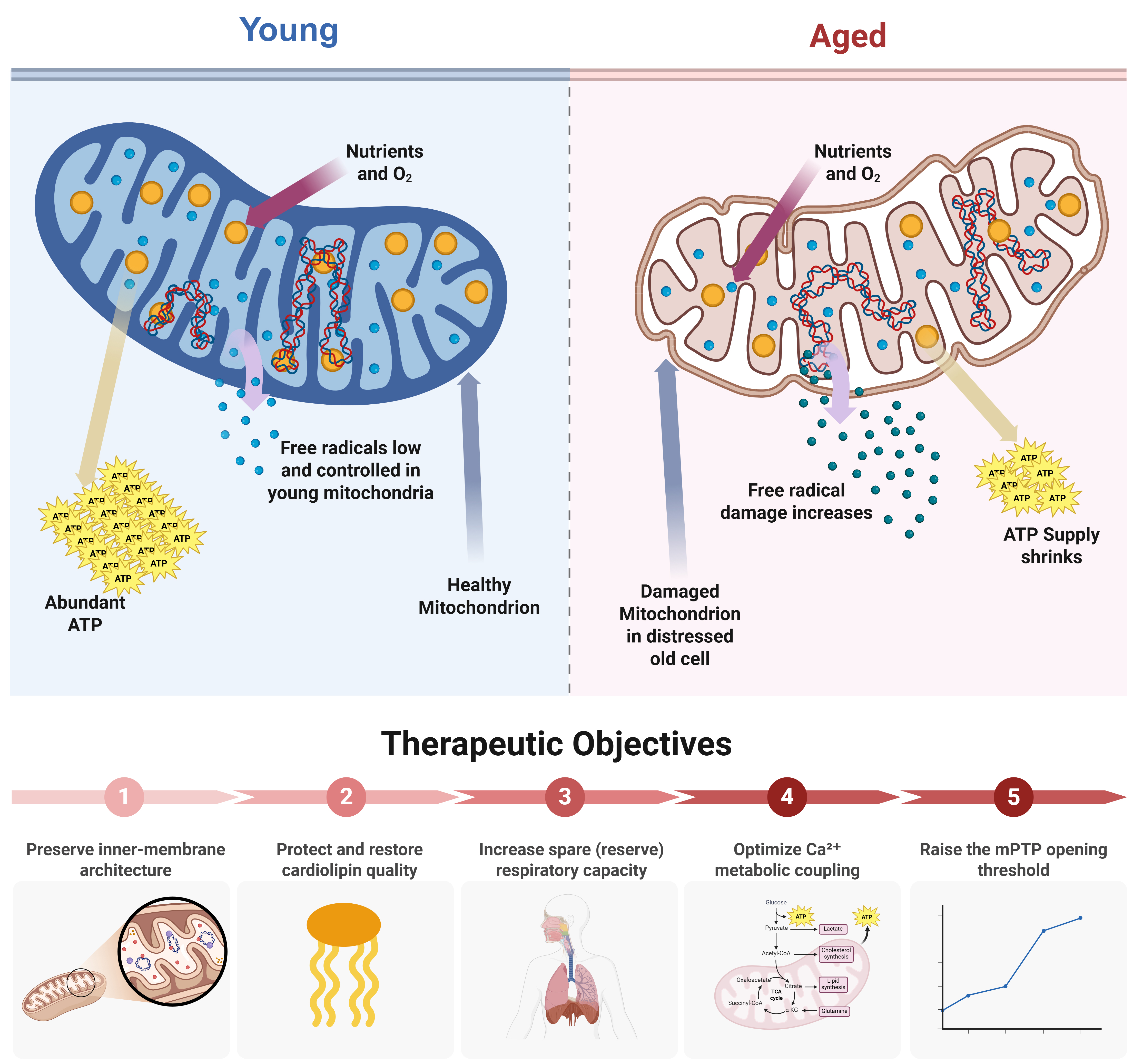
Figure 4. Structural and functional remodeling of mitochondria with aging and corresponding therapeutic objectives. (Created in BioRender. Ahmad D (2026) https://BioRender.com/p7qla0o). ATP: Adenosine triphosphate; TCA: tricarboxylic acid; mPTP: mitochondrial permeability transition pore; α-KG:
Mitochondrial-targeted clinical trials: lessons from past antioxidant failures
Early clinical efforts to target mitochondrial dysfunction in cardiovascular disease relied largely on systemic antioxidant supplementation, including vitamins C and E or beta-carotene. Despite strong mechanistic rationale and promising preclinical findings, large randomized trials generally failed to demonstrate cardiovascular benefit. One major reason for this failure is now understood to be the lack of mitochondrial specificity. Conventional antioxidants distribute widely throughout the cell and circulation and do not effectively accumulate within mitochondria, where the majority of ROS involved in cardiovascular aging are generated. Moreover, indiscriminate suppression of ROS may disrupt physiologic redox signaling that is necessary for adaptive stress responses, endothelial signaling, and metabolic regulation[95,96].
In contrast, newer therapeutic strategies specifically target mitochondrial biology and therefore address the underlying mechanisms of mitochondrial network dysfunction. Mitochondria-targeted antioxidants such as MitoQ are designed to accumulate within the mitochondrial matrix via membrane-potential-dependent uptake, allowing more direct modulation of mitochondrial redox balance[97]. Similarly, the cardiolipin-interacting peptide elamipretide (SS-31) stabilizes IMM architecture and improves ETC efficiency, while interventions such as urolithin A enhance mitochondrial turnover by stimulating mitophagy pathways[9,98]. Early human trials of these mitochondria-targeted strategies have demonstrated improvements in vascular function, mitochondrial biomarkers, and functional performance in aging populations. These findings suggest that therapies that restore mitochondrial quality control and bioenergetic reserve, rather than broadly suppressing oxidative stress, may provide a more effective translational pathway for improving cardiovascular resilience during aging.
UNDERSTANDING THE NETWORK IN MITOCHONDRIA: KEYS TO YOUTHFUL ORGANELLES
Therapeutic objective: preserving mitochondrial reserve to sustain cardiac resilience
Youthful organelles can be operationally defined in cardiovascular terms by their capacity to sustain energetic flexibility, stress-responsive reserve, and quality-control competence rather than by maximal basal ATP production alone. The essential therapeutic goal is not to maximize basal ATP in resting myocardium, but to preserve the reserve capacity and stability margins that allow mitochondria to meet abrupt, beat-to-beat demand without triggering maladaptive signaling or injury. In the aging heart, vulnerability emerges when the energetic system approaches critical thresholds[99], including loss of spare respiratory capacity[100], increased electron leak and lipid peroxidation[101], impaired Ca2+-metabolic coupling[100], and a lowered barrier to permeability transition such that physiologic stresses (adrenergic drive, transient ischemia, pressure overload) precipitate disproportionate dysfunction[102]. Accordingly, a “young” mitochondrial state can be defined as one that maintains (i) high-flux oxidative phosphorylation with low leak[103], (ii) preserved inner-membrane architecture and lipid quality[104], (iii) competent quality-control turnover[46], and (iv) a wide operating window between adaptive Ca2+-stimulated metabolism, transient permeability, [Figure 4] and catastrophic mitochondrial permeability transition pore (mPTP) opening[105]. These intervention priorities span preservation of inner-membrane architecture, cardiolipin stabilization, enhancement of spare respiratory capacity, optimization of Ca2+-metabolic coupling, and elevation of the mPTP opening threshold [Figure 4].
Inner-membrane architecture as a determinant of high-flux efficiency
Youthful mitochondria support high-velocity energy transfer because their inner membranes maintain sharp cristae curvature and cardiolipin (CL)-rich microdomains that pack the ETC and ATP synthase into kinetically favorable assemblies. Rows of ATP synthase dimers sculpt the cristae ridge, locally retaining protons and shortening the diffusion path between proton pumps and F1Fo-ATP synthase[106,107]; OPA1 and mitochondrial contact site and cristae organizing system (MICOS) help preserve this architecture, which in turn stabilizes ATP synthase oligomers under load[108]. When this mesoscale order is intact, electron flux can be rapidly increased without excessive leak[109]. CL is central at each step: it binds complexes I, III, and IV and promotes their higher-order association into respiratory supercomplexes, reducing intercomplex diffusion barriers and minimizing electron slip and ROS[110]. With aging and oxidative stress, CL becomes peroxidized and partially depleted, cristae flatten, and supercomplexes destabilize. As a result, complex activities and carrier proteins [e.g., adenine nucleotide translocase (ANT)] lose CL-dependent support, raising resistance to proton pumping and lowering the ceiling of oxidative phosphorylation (OXPHOS) throughput even when basal ATP is maintained[111]. Experimental restoration of CL content or biophysical properties, or reinforcement of cristae curvature, correspondingly improves coupling efficiency and peak respiratory flux[9].
Reserve capacity and Ca2+-coupled metabolism define the stress-tolerance window
The earliest functional signature of this architectural erosion is loss of reserve (spare) respiratory capacity, the difference between maximal and basal oxygen consumption, rather than a fall in resting ATP. Reserve capacity integrates multiple control points spanning substrate delivery, NADH/reduced flavin adenine dinucleotide (FADH2) supply, electron transfer kinetics, proton pumping, and ATP synthase conductance; aging narrows this margin, so mitochondria appear competent at baseline yet fail during stress tests (adrenergic stimulation, ischemia-reperfusion, or uncoupler-driven demands for maximal flux)[99,112]. In cardiomyocytes, stress tolerance also depends on tight coupling of cytosolic Ca2+ signals to mitochondrial metabolism while remaining safely below the threshold for permeability transition[113]. Matrix Ca2+ uptake through the mitochondrial calcium uniporter (MCU) activates TCA-cycle dehydrogenases (including pyruvate dehydrogenase via pyruvate dehydrogenase phosphatase 1 (PDP1), NAD-isocitrate dehydrogenase, and 2-oxoglutarate dehydrogenase), thereby matching reducing-equivalent supply to contractile work on short timescales; impairment of this axis blunts workload-induced ATP augmentation[113]. With age, redox tone and microdomain organization shift in ways that allow Ca2+-driven stimulation to more readily co-activate ROS production and lower the threshold for mPTP opening, increasing susceptibility to ΔΨ collapse and injury during stress, even when resting energetics appear preserved[114]. Preserving CL integrity and cristae architecture, therefore, expands the operating window between adaptive Ca2+-stimulated flux and catastrophic permeability transition[108].
MITOCHONDRIA AS SENTINELS IN CARDIOVASCULAR AGING
Mitochondria act as sentinels in cardiovascular aging because they sit at the intersection of energy supply, redox control, and inflammatory triggering - three constraints that determine whether stress is resolved cleanly or converted into chronic dysfunction. In the heart, mitochondrial performance sets the ceiling for contractile reserve during adrenergic drive, pressure overload, or transient ischemia[115]. In the vasculature, mitochondrial redox signaling helps determine NO bioavailability and the endothelial inflammatory tone that governs perfusion and vascular homeostasis[32]. When these mitochondrial functions are preserved, the system retains a wide safety margin: routine fluctuations in workload are buffered without durable shifts in gene programs or tissue structure. With aging, that margin narrows[29]. The same stressors increasingly produce disproportionate oxidant signaling, impaired vasodilatory responses, and activation of sterile inflammatory pathways that then reinforce metabolic and vascular dysfunction[33].
Across cell types, the most influential mitochondrial network effects for cardiovascular aging are often best framed through the microvascular endothelium, including coronary microvascular endothelial cells, with consequences that extend across heart, skeletal muscle, brain, liver, adipose tissue, and immune compartments [Figure 5]. These cells regulate local blood flow distribution, barrier properties, leukocyte trafficking, and paracrine signaling that shape the tissue environment in which cardiomyocytes must operate[116]. Endothelial mitochondria do not primarily exist to sustain bulk ATP demand. Instead, they are potent redox and signaling nodes[32]. A recurring theme in vascular aging is that greater reliance on mitochondrial-derived oxidant signaling accompanies reduced NO-dependent, vasoprotective signaling, contributing to endothelial dysfunction and impaired microvascular control[33]. In practical terms, once microvascular endothelial signaling drifts toward higher oxidant tone and lower NO availability, the heart becomes more vulnerable even if cardiomyocyte energetics are only modestly impaired, because oxygen and substrate delivery become less well matched to demand[116].
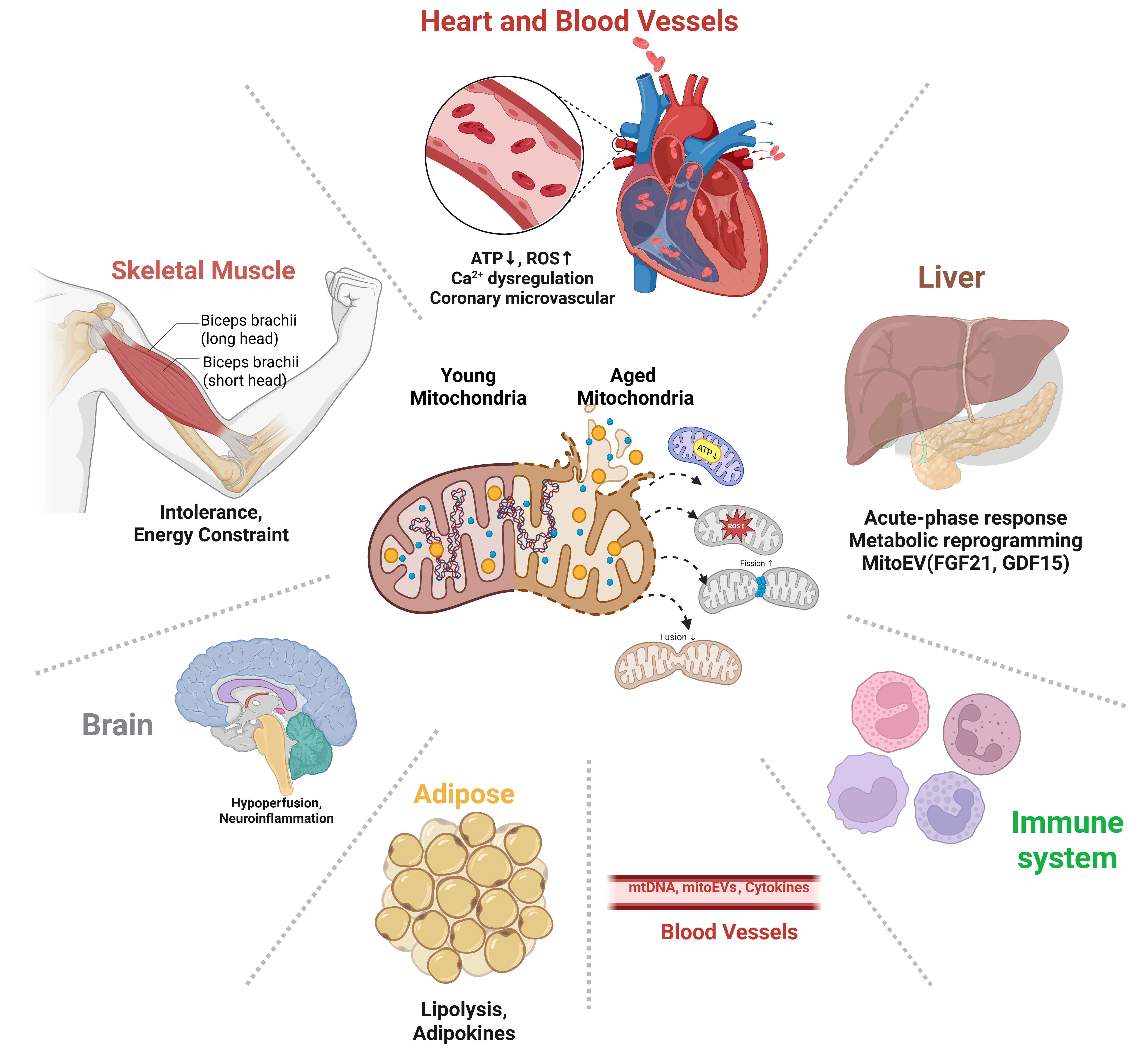
Figure 5. Organ-level consequences of mitochondrial aging and inter-organ communication networks. (Created in BioRender. Ahmad D (2026) https://BioRender.com/he5mt77). ATP: Adenosine triphosphate; ROS: reactive oxygen species; mitoEVs: mitochondrial extracellular vesicles; mtDNA: mitochondrial DNA; FGF21: fibroblast growth factor 21; GDF15: growth differentiation factor 15.
Cardiomyocytes remain essential to the story because their mitochondria supply the rapid, high-flux energy conversion required for beat-to-beat work. Aging-associated changes in mitochondrial structure and turnover, especially when they slow the removal of damaged organelles and the replacement of lost capacity, push cardiomyocytes closer to a threshold at which otherwise tolerable stresses provoke larger drops in energetic performance and larger redox perturbations[117]. Reviews of vascular and cardiac aging repeatedly emphasize that mitochondrial dynamics and mitophagy shape whether stress leaves behind a persistent population of dysfunctional mitochondria that continues to generate maladaptive signals rather than allowing the system to return to baseline after the inciting event[118].
A key mechanism linking mitochondrial injury to chronic cardiovascular inflammation is mislocalized mtDNA. When mitochondrial membranes or nucleoids are compromised, mtDNA can appear in the cytosol and engage innate immune DNA-sensing pathways such as cGAS-STING, which drives inflammatory transcriptional programs implicated across multiple cardiovascular disease settings[119]. This provides a direct route by which mitochondrial damage can shift both endothelial cells and cardiomyocytes toward a self-sustaining inflammatory state without infection, especially when aging-related declines in lysosomal clearance allow damaged mitochondrial material to persist[120].
These mechanisms have led to the emerging concept of mitoinflammation, which refers to sterile inflammation driven by mitochondrial damage and the release of mitochondrial danger-associated molecular patterns. Because mitochondria retain bacterial evolutionary characteristics, including circular DNA enriched in unmethylated CpG motifs and cardiolipin-containing membranes, mitochondrial components released during cellular stress are readily recognized by innate immune sensors[121,122]. Cytosolic mtDNA can activate the cGAS-STING signaling, which triggers interferon responses and NF-κB-dependent inflammatory transcription. In parallel, mitochondrial damage can promote activation of the Nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome through mechanisms that involve mitochondrial ROS generation, potassium efflux, and oxidized mtDNA signaling[123]. In the aging heart, persistent mitochondrial dysfunction therefore converts metabolic stress into chronic inflammatory signaling and reinforces the systemic low-grade inflammatory state known as inflammaging. This mitochondrial immune interface provides a mechanistic link between declining bioenergetic function and age-associated cardiovascular remodeling, including endothelial dysfunction, fibrosis, and microvascular inflammation[119].
When mitochondrial stress is prolonged, the response also extends beyond the local tissue through circulating stress-associated factors, often discussed as mitokines[124]. FGF21 and GDF15 are repeatedly described as factors induced downstream of mitochondrial dysfunction and integrated stress response signaling and can coordinate organism-level adaptations in metabolism and energy balance[80]. Sustained elevation is therefore better interpreted as a marker of unresolved mitochondrial strain than as a purely beneficial compensatory signal.
ENDOTHELIAL AND MICROVASCULAR MITOCHONDRIA AS GATEKEEPERS OF CARDIAC RESILIENCE
Cardiac performance depends on tight coupling between perfusion and energy use, and converging evidence places the earliest failure in the coronary microcirculation, where endothelial and perivascular mitochondria set tone, flow reserve, and nutrient delivery. In HFpEF, impaired coronary flow reserve and reduced myocardial perfusion reserve identify a clinically meaningful phenotype even when epicardial arteries are unobstructed, consistent with a small-vessel-first pathogenesis that precedes chamber failure and amplifies myocardial energetic stress[125]. Endothelial mitochondrial dysfunction contributes by reducing oxidative phosphorylation flexibility and increasing ROS, which in turn promotes endothelial nitric oxide synthase (eNOS) uncoupling, loss of NO bioavailability, and vasomotor stiffness. Aging and cardiometabolic risk further deepen this redox shift, increasing superoxide and peroxynitrite that oxidize tetrahydrobiopterin and critical eNOS thiols, thereby converting eNOS from a NO source to a superoxide source[126]. In parallel, the endothelial glycocalyx thins and sheds under inflammatory and hemodynamic stress, blunting shear sensing and flow-mediated dilation and increasing leukocyte adhesion and albumin leak[127]. Multi-cohort and mechanistic studies in HFpEF show higher circulating markers of glycocalyx degradation, such as perlecan fragments[128], and link endothelial injury to reduced coronary flow reserve and worse outcomes[129]. In parallel, endothelial mitochondrial ROS-driven NO deficiency has direct downstream consequences for the myocardium. Loss of endothelial NO bioavailability suppresses soluble guanylate cyclase/cyclic guanosine monophosphate/protein kinase G (sGC/cGMP/PKG) signaling in adjacent cardiomyocytes, favoring titin hypophosphorylation, increased passive tension, and impaired diastolic compliance, while chronic endothelial inflammatory activation promotes fibroblast activation and interstitial matrix deposition. Through this endothelial-to-myocardial signaling axis, microvascular mitochondrial dysfunction can be translated into myocardial stiffness and impaired relaxation, two central features of HFpEF [Figure 6].
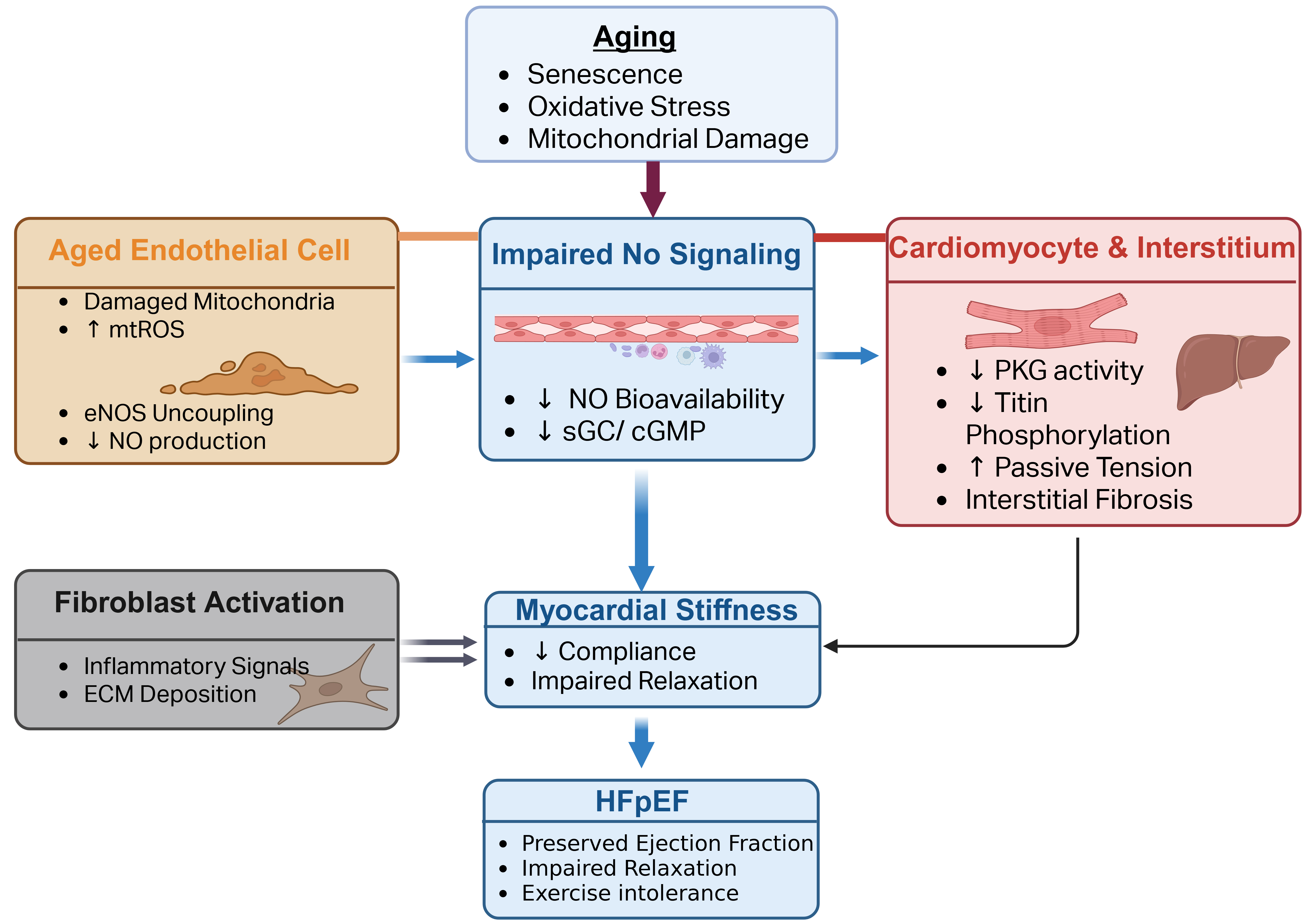
Figure 6. Endothelial to Myocardial Signaling in Cardiovascular Aging: Mitochondrial ROS, NO Deficiency, and Myocardial Stiffness Driving HFpEF (Created in BioRender. Shila TA (2026) https://BioRender.com/zlp092e). HFpEF: Heart failure with preserved ejection fraction; ROS: reactive oxygen species; eNOS: endothelial nitric oxide synthase; sGC: soluble guanylate cyclase; cGMP: cyclic guanosine monophosphate; PKG: protein kinase G; mtROS: mitochondrial reactive oxygen species; NO: nitric oxide; ECM: extracellular matrix.
Pericytes provide a second control layer that becomes critical as endothelial mitochondria age. These mural cells stabilize capillaries[130], shape barrier properties[131], and regulate capillary diameter through a contractile apparatus that responds to metabolic and redox cues[132], thereby tuning local resistance and flow heterogeneity across the capillary network. Comparative work in brain and heart shows that pericyte-mediated capillary constriction can limit downstream perfusion after ischemia and likely in other pathological states, while experimental loss or phenotypic drift of cardiac pericytes initiates microvascular dysfunction, heightens endothelial inflammation, and contributes to diastolic dysfunction[133]. High-resolution studies and reviews demonstrate that pericytes influence angiogenesis, basement membrane composition, and capillary recruitment, and that their depletion or hypercontractility increases flow heterogeneity, lengthens oxygen diffusion distances, and reduces reserve during stress despite normal upstream conductance[134]. In clinical and preclinical contexts, these microcirculatory changes increase ischemia susceptibility and erode energetic reserve, providing a mechanistic bridge between capillary level dynamics and the exercise intolerance and demand ischemia that characterize HFpEF.
From a forward-looking perspective, endothelial aging should be conceptualized as a primary disease-modifying process that determines whether systemic cardiometabolic stress is buffered within the microcirculation or propagated to the myocardium as chronic energetic strain. A major priority for the field is mechanistic stratification of endothelial dysfunction into definable mitochondrial-linked failure modes[135], including loss of mitochondrial redox control with eNOS uncoupling[126], impaired mitochondrial turnover and stress tolerance[136], disruption of NAD+-dependent signaling programs[137], and mtDNA-associated innate immune activation, each of which is predicted to produce distinct signatures in flow reserve[138], barrier permeability, and inflammatory activation[139]. In parallel, improved clinical phenotyping is needed to connect microvascular pathobiology to outcomes using integrated measures of coronary flow reserve and perfusion reserve alongside circulating or imaging-based indices of endothelial injury, including glycocalyx degradation and mitochondrial stress biomarkers[129]. This framework motivates a translational agenda for the next section in which candidate therapies are evaluated not only by hemodynamic endpoints, but also by their capacity to restore endothelial mitochondrial homeostasis, preserve glycocalyx mechanotransduction, and normalize pericyte-dependent capillary recruitment, thereby re-establishing perfusion-metabolism coupling before irreversible myocardial remodeling and HFpEF progression occur.
HUMAN EVIDENCE AND MEASUREMENT: DEFINING MITOCHONDRIAL AGE
Defining “mitochondrial age” in humans is most informative when anchored in functional reserve and stress responses rather than static measures of mitochondrial content. At the tissue level, dynamic phosphorus magnetic resonance spectroscopy (MRS) can quantify post-exercise phosphocreatine recovery kinetics, which serve as an in vivo index of oxidative phosphorylation capacity because phosphocreatine resynthesis is driven by mitochondrial ATP production; slower recovery reflects reduced oxidative capacity and a more aged bioenergetic phenotype even when resting ATP appears preserved[140].
At the cellular scale, optical metabolic imaging approaches such as fluorescence lifetime imaging of endogenous NAD(P)H (reduced nicotinamide adenine dinucleotide phosphate and reduced nicotinamide adenine dinucleotide) and flavin adenine dinucleotide (FAD) can report enzyme engagement and redox state by distinguishing bound and free lifetime components and by tracking redox ratio shifts across cells and tissues, enabling label-free detection of metabolic state changes in translational samples[141]. In blood, peripheral immune-cell bioenergetic profiling offers an accessible “systemic stress test” of mitochondrial resilience, including reserve and spare respiratory capacity measured by respirometric methods; this approach is increasingly used to relate mitochondrial function to aging phenotypes, frailty, and cardiometabolic risk[142].
Circulating cell-free mtDNA provides an additional minimally invasive window into mitochondrial damage signaling and sterile inflammation in cardiovascular contexts, but it requires careful interpretation because levels are sensitive to preanalytical handling and to the specific physiological or disease context in which mitochondrial material is released[143]. Human studies also emphasize key modifiers that must be incorporated into trial design to improve signal-to-noise and interpretability, including tissue specificity of mitochondrial aging, sex-related differences in mitochondrial bioenergetics, and circadian regulation of metabolism and vascular function[144-146].
For clinical translation, measurement should follow a mechanism-anchored playbook. Endpoints should be paired to the hypothesized therapeutic lever and captured under standardized conditions, with core-laboratory adjudication when feasible. For endothelial coupling, brachial artery flow-mediated dilation remains a widely used, shear-dependent, nitric-oxide-linked measure with established expert consensus recommendations that improve reliability when protocols are standardized[147]. For mitochondrial functional reserve, practical trial panels can combine (i) phosphocreatine recovery kinetics from 31P MRS as a tissue oxidative-capacity readout, (ii) peripheral blood cell spare respiratory capacity as a systemic resilience metric, (iii) NAD(P)H and FAD lifetime metrics for cellular redox and enzyme engagement, and (iv) circulating cell-free mtDNA as a damage and inflammatory-signal readout, with all components normalized to age and sex and measured at consistent circadian time points[140,143,147-149]. An optional vascular-coupling subscore can integrate endothelial function with perfusion or coronary flow reserve, where microvascular supply-demand matching is central.
Patient enrichment should similarly be mechanism-driven. Early phenotypes in which mitochondrial and microvascular lesions dominate, such as HFpEF with exercise-limited hemodynamics or microvascular angina with impaired perfusion reserve despite unobstructed epicardial arteries, are plausible settings in which mitochondria-targeted therapies may yield clearer functional signals than in advanced remodeling states.
NEW PERSPECTIVES ON IMPROVING MITOCHONDRIAL YOUTHFULNESS AND THE MEASUREMENT PRECISION
A coherent picture has begun to emerge in which mitochondrial quality control is a modifiable driver of clinical aging, and the features of a minimum effective rejuvenation strategy can be inferred from human and translational data. Restoration of inner-membrane quality that supports cristae curvature and cardiolipin binding appears central, because cardiolipin stabilizes respiratory supercomplexes and improves electron transfer efficiency[9,150]. In failing human hearts, the cardiolipin-interacting tetrapeptide elamipretide improved complex I and IV activities, supercomplex-linked respiration, and overall oxygen flux in ex vivo assays of biopsy tissue[9], indicating that relatively small biophysical shifts at the membrane can translate into measurable gains in flux capacity that matter under stress. Parallel evidence supports the mitophagy arm of quality control. In randomized trials in middle-aged and older adults, supplementation with urolithin A increased performance and endurance while improving biomarkers of mitochondrial health that reflect enhanced mitophagy, establishing target engagement and functional benefit in humans without disease-stage confounders[90]. At the failure end of the spectrum, age-dependent sensitization of the mPTP lowers the threshold for stress-induced loss of membrane potential in the heart, linking impaired quality control to vulnerability during ischemia-reperfusion or adrenergic load. Raising this threshold is therefore a plausible criterion for effective rejuvenation and is more closely linked to functional outcomes than static content measures[151]. These studies suggest that a minimally effective package will likely include membrane-level remediation that restores cardiolipin-supported architecture, measurable increases in mitophagy with preserved lysosomal clearance, and a demonstrable upward shift in the permeability transition threshold, with clinical translation judged by gains in stress-tested reserve rather than resting surrogates. A summary of mitochondria-targeted therapeutic strategies, their mechanistic targets, and current clinical status is provided in Table 2.
Mitochondria-targeted therapeutic strategies in cardiovascular aging
| Intervention | Mechanistic target | Primary mito lever (energy/quality/signaling) | Model/study type | Key cardiovascular findings | Clinical status |
| MitoQ (mitoquinone)[155] | Mitochondrial ROS modulation | Signaling/Energy | Randomized controlled trial in healthy older adults | Improved brachial artery flow-mediated dilation; reduced aortic stiffness | Completed human RCT |
| Elamipretide (SS-31)[9] | Cardiolipin stabilization; cristae integrity | Energy/Quality | Ex vivo failing human myocardium; Barth syndrome trials | Improved complex I/IV activity and supercomplex respiration; enhanced mitochondrial oxygen flux | FDA accelerated approval (Barth syndrome) |
| Urolithin A[90,98] | Mitophagy activation | Quality | Randomized placebo-controlled trials in middle-aged and older adults | Improved muscle endurance; increased biomarkers of mitochondrial health | Human RCT completed |
| Nicotinamide riboside (NR)[91] | NAD+ repletion; sirtuin activation | Energy/Signaling | Human pilot RCT in hypertensive middle-aged/older adults | Improved vascular parameters; enhanced metabolic resilience | Early-phase human trials |
| AMPK activators (exercise, pharmacologic)[156] | Energy sensing; mitochondrial biogenesis | Energy/Quality | Animal aging models; human exercise studies | Increased mitochondrial content and respiratory capacity; improved diastolic reserve | Established lifestyle therapy; pharmacologic agents under study |
| Time-restricted eating (TRE)[92] | Nutrient-sensing alignment; metabolic stress modulation | Energy/Signaling | Human interventional studies in older adults | Improved endothelial function; vascular coupling endpoints under evaluation | Ongoing clinical trials |
| SGLT2 inhibitors (e.g., empagliflozin)[94] | Improved mitochondrial redox state; endothelial bioenergetics | Energy/Signaling | Mechanistic human studies; diabetic mouse models; large CV outcome trials | Improved endothelial function; reduced oxidative stress; improved HF outcomes | FDA-approved for HF and diabetes |
| Mitophagy enhancers (e.g., BNIP3/NIX pathway targeting, experimental compounds)[157,158] | Selective mitochondrial clearance | Quality | Transgenic mouse models of impaired mitophagy | Restored mitochondrial turnover; improved cardiac stress tolerance | Preclinical stage |
| mPTP modulation strategies[159] | Increase permeability transition threshold | Quality/Energy | Aged rodent heart models | Reduced ischemia-reperfusion injury; improved stress resilience | Preclinical/experimental |
Coordination across the myocardial syncytium has been most clearly described at the endothelial-cardiomyocyte interface, and the literature points to shared signals that can be mapped and perturbed. Endothelial mitochondria help maintain NO bioavailability; with aging, redox shifts, and tetrahydrobiopterin oxidation promote eNOS synthase uncoupling, thereby reducing flow-mediated dilation and limiting perfusion reserve during stress[152]. Communication is not confined to classical mediators. Mitochondrial content and signals also traffic between cell types in EVs that can carry mtDNA and proteins. This vesicle export increases when lysosomal clearance is constrained, providing a route by which local quality-control bottlenecks propagate inflammatory tone and metabolic cues across the vessel wall and into the interstitium[153]. Systemic coordination is further suggested by mitochondrial stress-induced secreted factors, including the mitokines FGF21 and GDF15, which are transcriptionally induced by integrated stress responses and secreted by stressed tissues to couple organelle dysfunction to whole-body metabolic adaptation[80]. By contrast, mitochondria are not considered a canonical secretory source of cytokines; mitochondrial damage more commonly amplifies cytokine production indirectly through the release of mitochondrial danger-associated molecular patterns (DAMPs), such as mtDNA, that engage innate immune sensors[119]. Mitokines are mitochondrial stress-induced secreted factors, and the term is functional rather than taxonomic; it overlaps with, but is not synonymous with, cytokines, as shown by GDF15, a stress-induced cytokine of the transforming growth factor-beta (TGF-β) superfamily, and FGF21, an endocrine metabolic hormone, both of which rise in response to mitochondrial dysfunction[124]. Mitochondria are not generally thought to secrete cytokines directly; instead, mitochondrial stress and damage promote cytokine production by exporting mitochondrial danger signals such as mtDNA that activate innate immune pathways including cGAS-STING and the NLRP3 inflammasome[154]. A working model therefore envisions endothelial mitochondria as gatekeepers of shear sensing and NO signaling that communicate with cardiomyocytes through redox and calcium microdomains, through metabolite exchange such as lactate and ketone use, and through vesicle traffic and secreted-factor signaling, including cytokines and mitokines, which together reflect sustained stress when local repair falls behind.
Measurements of mitochondrial aging should be investigated using standardized, mechanism-anchored frameworks. Progress will depend on reporting standards that allow mitochondrial endpoints to be compared and pooled across cardiovascular trials. For energy, 31P MRS of phosphocreatine recovery after brief exercise is reproducible, correlates with in vitro oxidative capacity, and should be paired with standardized measures of oxygen consumption (VO2) kinetics during ramp exercise and with myocardial perfusion or coronary flow reserve obtained under defined shear or adrenergic stimuli. For endothelial function, consensus statements on flow-mediated dilation and related measures provide procedures, calibration steps, and diagnostic thresholds that can be harmonized across sites; adherence to these guidance documents reduces variability and improves interpretability of shear-dependent endpoints directly tied to endothelial mitochondrial state through NO biology. For quality, trial reports should include validated proxies of cardiolipin status when tissue is available, high-resolution respirometry signatures that separate leak, coupling, and spare capacity, and dynamic markers of mitophagy and lysosomal flux. Interpretation should also account for the possibility that release of mitochondrial material in large EVs reflects downstream clearance constraints. For signaling, preanalytical handling and timing for circulating cell-free mtDNA and redox and acetylation panels should be specified, and endothelial outcomes should be adjudicated by a core laboratory to permit cross-trial synthesis. With these standards, the field will be better positioned to quantify the smallest effective quality-control rejuvenation that improves patient function and to test coordinated interventions across endothelial and myocardial compartments.
CONCLUSION
Cardiovascular aging can be understood as a progressive failure of a mitochondrial network that spans the myocardium, endothelium, and microvasculature. Across the literature reviewed here, three linked domains consistently account for this loss of resilience. Energy declines when spare respiratory capacity contracts and perfusion-metabolism coupling become unreliable under load, even while resting ATP remains near normal. Quality falters when cristae and cardiolipin architecture deteriorate, when fission-fusion balance drifts, and when mitophagy and lysosomal clearance cannot keep pace with damage. Signaling becomes maladaptive when mtDNA and peptides trigger innate immunity through cGAS-STING, when compartment-specific NAD+-SIRT control slips, and when stress-induced cytokines and mitokines chronically reinforce cellular stress programs. These processes interact most visibly in endothelial and microvascular territories that gate flow and barrier function, thereby setting an early ceiling on oxygen delivery that is then inherited by cardiomyocytes during exercise or ischemia. Viewing the problem through this network lens replaces a single-lesion narrative with a framework of tractable nodes and links that can be measured, perturbed, and followed over time. Translation should proceed by measuring what matters, intervening where the earliest failures occur, and defining success by restored function under stress. The proposed Mitochondrial Functional Age panel integrates dynamic energy metrics, markers of organelle quality and turnover, and indices of signaling tone, while incorporating circadian timing and site standards to support comparability. Such a composite can enrich trials for early phenotypes characterized by perfusion-metabolism uncoupling, and it can anchor go-no-go decisions to mechanisms rather than to distant outcomes alone. Interventions that increase reserve capacity, stabilize cristae and cardiolipin architecture, and normalize mitophagy-lysosome flux, together with endothelial-targeted strategies that recover NO biology and glycocalyx integrity, are most likely to improve clinically relevant endpoints such as exercise hemodynamics, diastolic reserve, and recovery kinetics. Genetic and nucleic acid-based approaches remain promising but will require careful attention to delivery, safety, and dose control. If these elements are adopted, mitochondrial youthfulness can become a practical therapeutic objective rather than a merely descriptive label. A field that routinely quantifies reserve, quality control, and signaling, targets endothelial and intermyofibrillar microdomains, and judges success by improved coupling of perfusion to work will be positioned to convert mechanism into medicine. The expected payoff is not only the delayed onset of overt disease but also measurable gains in everyday cardiovascular function in older adults.
DECLARATIONS
Acknowledgement
The graphical abstract was created in BioRender. Ahmad D (2026) https://BioRender.com/dwtuhm3.
Authors’ contributions
Conceived, synthesized and edited the manuscript: Wang Y, Zhao J
Drafted, prepared figures, edited, and revised the manuscript: Shila TA, Ahmad D, Tan H, Wang B, Zhang J
Availability of data and material
Not applicable
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (OpenAI; version GPT-5.2, released 2025-12-11) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by awards from the National Institutes of Health (RO1HL123404, RO1HL-96686, Xin-Liang Ma/Wang Y, MPI; RO1HL-157495, RO1HL158612, RO1HL173090, Wang Y), the American Heart Association (20TPA1293530, Wang Y; 25PRE1377191, Zhang J) and the Engineering School of UAB Innovative Fellowship Award in 2024.
Conflicts of interest
Wang Y is an Editorial Board Member of The Journal of Cardiovascular Aging. Wang Y was not involved in any steps of the editorial process, notably including reviewers’ selection, manuscript handling, or decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Dai D, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109-24.
2. Ciumărnean L, Milaciu MV, Negrean V, et al. Cardiovascular risk factors and physical activity for the prevention of cardiovascular diseases in the elderly. Int J Environ Res Public Health. 2021;19:207.
3. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016;118:1593-611.
4. Li Y, Jin X, Li D, et al. New insights into vascular aging: emerging role of mitochondria function. Biomed Pharmacother. 2022;156:113954.
5. Ding W, Chen J, Zhao L, Wu S, Chen X, Chen H. Mitochondrial DNA leakage triggers inflammation in age-related cardiovascular diseases. Front Cell Dev Biol. 2024;12:1287447.
6. Brown DA, Perry JB, Allen ME, et al. Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2016;14:238-50.
7. Aon MA, Tocchetti CG, Bhatt N, Paolocci N, Cortassa S. Protective mechanisms of mitochondria and heart function in diabetes. Antioxid Redox Signal. 2015;22:1563-86.
8. Szeto HH, Liu S. Cardiolipin-targeted peptides rejuvenate mitochondrial function, remodel mitochondria, and promote tissue regeneration during aging. Arch Biochem Biophys. 2018;660:137-48.
9. Chatfield KC, Sparagna GC, Chau S, et al. Elamipretide improves mitochondrial function in the failing human heart. JACC Basic Transl Sci. 2019;4:147-57.
10. Ikeda Y, Sciarretta S, Nagarajan N, et al. New insights into the role of mitochondrial dynamics and autophagy during oxidative stress and aging in the heart. Oxid Med Cell Longev. 2014;2014:1-13.
11. Scheffer DDL, Garcia AA, Lee L, Mochly-rosen D, Ferreira JCB. Mitochondrial fusion, fission, and mitophagy in cardiac diseases: challenges and therapeutic opportunities. Antioxid Redox Signal. 2022;36:844-63.
12. Kirkman DL, Robinson AT, Rossman MJ, Seals DR, Edwards DG. Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2021;320:H2080-100.
13. Rouhi L, Gurha P, Marian AJ. The CGAS-STING1 pathway as a mediator of innate immune response in cardiovascular disease. JACC Asia. 2025;5:516-27.
14. López-otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243-78.
15. Lozhkin A, Vendrov AE, Ramos-mondragón R, et al. Mitochondrial oxidative stress contributes to diastolic dysfunction through impaired mitochondrial dynamics. Redox Biology. 2022;57:102474.
16. Chakravarti D, Labella KA, Depinho RA. Telomeres: history, health, and hallmarks of aging. Cell. 2021;184:306-22.
17. Guo J, Huang X, Dou L, et al. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Sig Transduct Target Ther. 2022;7:391.
18. Ahmad D, Shila TA, Yang Y, Tan H, Wang Y. Current diagnostic modalities for detecting myocardial relaxation function in diabetes-associated heart failure or heart failure with preserved ejection fraction. Curr Opin Physiol. 2025;46:100863.
19. Viloria MAD, Li Q, Lu W, et al. Effect of exercise training on cardiac mitochondrial respiration, biogenesis, dynamics, and mitophagy in ischemic heart disease. Front Cardiovasc Med. 2022;9:949744.
20. Nair S, Ren J. Autophagy and cardiovascular aging: lesson learned from rapamycin. Cell Cycle. 2014;11:2092-9.
21. Barcena ML, Aslam M, Norman K, Ott C, Ladilov Y. Role of AMPK and sirtuins in aging heart: basic and translational aspects. Aging Dis. 2024;16:3335-60.
22. Wu S, Zou M. AMPK, mitochondrial function, and cardiovascular disease. Int J Mol Sci. 2020;21:4987.
23. Faitg J, D’amico D, Rinsch C, Singh A. Mitophagy activation by urolithin a to target muscle aging. Calcif Tissue Int. 2023;114:53-9.
24. Glancy B, Kim Y, Katti P, Willingham TB. The functional impact of mitochondrial structure across subcellular scales. Front Physiol. 2020;11:541040.
25. Kurz FT, Aon MA, O’rourke B, Armoundas AA. Cardiac mitochondria exhibit dynamic functional clustering. Front Physiol. 2014;5:1-8.
26. Jiang Z, Shen T, Huynh H, Fang X, Han Z, Ouyang K. Cardiolipin regulates mitochondrial ultrastructure and function in mammalian cells. Genes. 2022;13:1889.
27. Kurz FT, Derungs T, Aon MA, O’rourke B, Armoundas AA. Mitochondrial networks in cardiac myocytes reveal dynamic coupling behavior. Biophys J. 2015;108:1922-33.
28. Aon MA, Cortassa S, O’rourke B. The fundamental organization of cardiac mitochondria as a network of coupled oscillators. Biophys J. 2006;91:4317-27.
29. Ali MA, Gioscia-ryan R, Yang D, Sutton NR, Tyrrell DJ. Cardiovascular aging: spotlight on mitochondria. Am J Physiol Heart Circ Physiol. 2024;326:H317-33.
30. Padrão AI, Ferreira RM, Vitorino R, et al. OXPHOS susceptibility to oxidative modifications: the role of heart mitochondrial subcellular location. Biochim Biophys Acta Bioenerg. 2011;1807:1106-13.
31. Kasumov T, Dabkowski ER, Shekar KC, et al. Assessment of cardiac proteome dynamics with heavy water: slower protein synthesis rates in interfibrillar than subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2013;304:H1201-14.
32. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013;112:1171-88.
33. Tracy EP, Hughes W, Beare JE, Rowe G, Beyer A, Leblanc AJ. Aging-induced impairment of vascular function: mitochondrial redox contributions and physiological/clinical implications. Antioxid Redox Signal. 2021;35:974-1015.
34. Xu X, Pang Y, Fan X. Mitochondria in oxidative stress, inflammation and aging: from mechanisms to therapeutic advances. Sig Transduct Target Ther. 2025;10:190.
35. Chen J, Zhong J, Wang L, Chen Y. Mitochondrial transfer in cardiovascular disease: from mechanisms to therapeutic implications. Front Cardiovasc Med. 2021;8:771298.
36. Iorio R, Petricca S, Di Emidio G, Falone S, Tatone C. Mitochondrial extracellular vesicles (mitoEVs): emerging mediators of cell-to-cell communication in health, aging and age-related diseases. Ageing Res Rev. 2024;101:102522.
37. Wu S, Yang T, Ma M, et al. Extracellular vesicles meet mitochondria: potential roles in regenerative medicine. Pharmacol Res. 2024;206:107307.
38. Zhang Y, Yu Z, Jiang D, et al. iPSC-MSCs with high intrinsic MIRO1 and sensitivity to TNF-α yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Reports. 2016;7:749-63.
39. Liu K, Ji K, Guo L, et al. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res. 2014;92:10-8.
40. Manickam DS, Pinky PP, Khare P. Extracellular vesicle-mediated mitochondria delivery: premise and promise. J Cereb Blood Flow Metab. 2025;46:306-21.
41. Nicolás-Ávila JA, Lechuga-vieco AV, Esteban-martínez L, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. 2020;183:94-109.e23.
42. O’brien CG, Ozen MO, Ikeda G, et al. Mitochondria-rich extracellular vesicles rescue patient-specific cardiomyocytes from doxorubicin injury: insights into the SENECA trial. JACC CardioOncology. 2021;3:428-40.
43. Ikeda G, Santoso MR, Tada Y, et al. Mitochondria-rich extracellular vesicles from autologous stem cell-derived cardiomyocytes restore energetics of ischemic myocardium. J Am Coll Cardiol. 2021;77:1073-88.
44. Mone P, Agyapong ED, Morciano G, et al. Dysfunctional mitochondria elicit bioenergetic decline in the aged heart. J Cardiovasc Aging. 2024;4:13.
45. Sansbury BE, Jones SP, Riggs DW, Darley-usmar VM, Hill BG. Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chem Biol Interact. 2011;191:288-95.
46. Liang WJ, Gustafsson ÅB. The aging heart: mitophagy at the center of rejuvenation. Front Cardiovasc Med. 2020;7:18.
47. Gabillard-lefort C, Thibault T, Lenaers G, Wiesner RJ, Mialet-perez J, Baris OR. Heart of the matter: mitochondrial dynamics and genome alterations in cardiac aging. Mech Ageing Dev. 2025;224:112044.
48. Molina-Riquelme I, Barrientos G, Breitsprecher L, et al. Multi-scale mitochondrial cristae remodeling links Opa1 downregulation to reduced OXPHOS capacity in aged hearts. bioRxiv 2025. https://www.biorxiv.org/content/10.1101/2025.04.01.644555v1.abstract (accessed 2026-05-12).
49. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: molecular and pharmacological aspects. Cells. 2019;8:728.
50. Kane AE, Sinclair DA. Sirtuins and NAD+ in the development and treatment of metabolic and cardiovascular diseases. Circ Res. 2018;123:868-85.
51. Yuan Y, Liang B, Liu X, et al. Targeting NAD+: is it a common strategy to delay heart aging? Cell Death Discov. 2022;8:230.
52. Zong Y, Li H, Liao P, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Sig Transduct Target Ther. 2024;9:124.
53. Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Sig Transduct Target Ther. 2023;8:333.
54. Hinton A, Claypool SM, Neikirk K, et al. Mitochondrial structure and function in human heart failure. Circ Res. 2024;135:372-96.
55. Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 2015;116:1477-90.
56. Xie S, Xu S, Deng W, Tang Q. Metabolic landscape in cardiac aging: insights into molecular biology and therapeutic implications. Sig Transduct Target Ther. 2023;8:114.
58. Shirihai OS, Song M, Dorn GW. How mitochondrial dynamism orchestrates mitophagy. Circ Res. 2015;116:1835-49.
59. Ajoolabady A, Aslkhodapasandhokmabad H, Aghanejad A, Zhang Y, Ren J. Mitophagy receptors and mediators: therapeutic targets in the management of cardiovascular ageing. Ageing Res Rev. 2020;62:101129.
60. Zhang R, Krigman J, Luo H, Ozgen S, Yang M, Sun N. Mitophagy in cardiovascular homeostasis. Mech Ageing Dev. 2020;188:111245.
61. Lampert MA, Orogo AM, Najor RH, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019;15:1182-98.
62. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res. 2016;118:1563-76.
63. Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E. Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ Res. 2012;110:1125-38.
64. Aman Y, Schmauck-medina T, Hansen M, et al. Autophagy in healthy Aging Dis. Nat Aging. 2021;1:634-50.
65. Zhang C, Lin S. AMPK promotes autophagy by facilitating mitochondrial fission. Cell Metab. 2016;23:399-401.
66. Ikeda Y, Shirakabe A, Maejima Y, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264-78.
67. Trouillard M, Meunier B, Rappaport F. Questioning the functional relevance of mitochondrial supercomplexes by time-resolved analysis of the respiratory chain. Proc Natl Acad Sci U S A. 2011;108:E1027-34.
69. Devin A, Rigoulet M. Mechanisms of mitochondrial response to variations in energy demand in eukaryotic cells. Am J Physiol Cell Physiol. 2007;292:C52-8.
70. Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959-73.
71. Simmen T, Tagaya M. Organelle communication at membrane contact sites (MCS): from curiosity to center stage in cell biology and biomedical research. In: Tagaya M, Simmen T, Editors. Organelle Contact Sites. Singapore: Springer Singapore; 2017. pp. 1-12.
72. Szabadkai G, Bianchi K, Várnai P, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901-11.
73. Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, Mcbride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE. 2012;7:e52830.
74. Todkar K, Chikhi L, Desjardins V, El-mortada F, Pépin G, Germain M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat Commun. 2021;12:1971.
75. Schvartzman JM, Thompson CB, Finley LW. Metabolic regulation of chromatin modifications and gene expression. J Cell Biol. 2018;217:2247-59.
76. Fessler E, Eckl E, Schmitt S, et al. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature. 2020;579:433-7.
77. Oduro PK, Zheng X, Wei J, et al. The cGAS-STING signaling in cardiovascular and metabolic diseases: future novel target option for pharmacotherapy. Acta Pharm Sin B. 2022;12:50-75.
78. Stephens OR, Grant D, Frimel M, et al. Characterization and origins of cell-free mitochondria in healthy murine and human blood. Mitochondrion. 2020;54:102-12.
79. Nakahira K, Kyung S, Rogers AJ, et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med. 2013;10:e1001577.
80. Jena J, García-peña LM, Pereira RO. The roles of FGF21 and GDF15 in mediating the mitochondrial integrated stress response. Front Endocrinol. 2023;14:1264530.
81. Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells - partnering up with the immune system? Nat Rev Immunol. 2022;22:576-88.
82. Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal. 2011;15:1517-30.
83. Kempf S, Popp R, Naeem Z, et al. Pericyte-to-endothelial cell communication via tunneling nanotubes is disrupted by a diol of docosahexaenoic acid. Cells. 2024;13:1429.
84. Nguyen TT, Wei S, Nguyen TH, et al. Mitochondria-associated programmed cell death as a therapeutic target for age-related disease. Exp Mol Med. 2023;55:1595-619.
85. Burtscher J, Soltany A, Visavadiya NP, et al. Mitochondrial stress and mitokines in aging. Aging Cell. 2023;22:e13770.
86. Yu H, Liao K, Hu Y, et al. Role of the cGAS-STING pathway in aging-related endothelial dysfunction. Aging Dis. 2022;13:1901.
87. Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol. 2013;305:H1417-27.
88. Higashi Y, Kihara Y, Noma K. Endothelial dysfunction and hypertension in aging. Hypertens Res. 2012;35:1039-47.
89. Chang X, Li Y, Cai C, et al. Mitochondrial quality control mechanisms as molecular targets in diabetic heart. Metabolism. 2022;137:155313.
90. Liu S, D’amico D, Shankland E, et al. Effect of urolithin a supplementation on muscle endurance and mitochondrial health in older adults: a randomized clinical trial. JAMA Netw Open. 2022;5:e2144279.
91. Lin Y, Zeidan RS, Lapierre-nguyen S, et al. Nicotinamide riboside combined with exercise to treat hypertension in middle-aged and older adults: a pilot randomized clinical trial. GeroScience. 2025;47:6895-908.
92. Da C. Pinaffi-langley AC, Szarvas Z, Peterfi A, et al. Time-restricted eating for prevention of age-related vascular cognitive decline in older adults: a protocol for a single-arm open-label interventional trial. PLoS ONE. 2024;19:e0314871.
94. Mone P, Varzideh F, Jankauskas SS, et al. SGLT2 inhibition via empagliflozin improves endothelial function and reduces mitochondrial oxidative stress: insights from frail hypertensive and diabetic patients. Hypertension. 2022;79:1633-43.
95. Cook NR. A randomized factorial trial of vitamins c and e and beta carotene in the secondary prevention of cardiovascular events in women: results from the women’s antioxidant cardiovascular study. Arch Intern Med. 2007;167:1610.
96. Jiang Q, Yin J, Chen J, et al. Mitochondria-targeted antioxidants: a step towards disease treatment. Oxid Med Cell Longev. 2020;2020:1-18.
97. Graham D, Huynh NN, Hamilton CA, et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322-8.
98. Singh A, D’amico D, Andreux PA, et al. Urolithin a improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell Reports Medicine. 2022;3:100633.
99. Desler C, Hansen TL, Frederiksen JB, Marcker ML, Singh KK, Juel Rasmussen L. Is there a link between mitochondrial reserve respiratory capacity and aging? J Aging Res. 2012;2012:1-9.
100. Fernandez-sanz C, Ruiz-meana M, Miro-casas E, et al. Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 2014;5:e1573.
101. Moghaddas S, Hoppel CL, Lesnefsky EJ. Aging defect at the QO site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 2003;414:59-66.
102. Hofer T, Servais S, Seo AY, et al. Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mech Ageing Dev. 2009;130:297-307.
103. Wikström M, Springett R. Thermodynamic efficiency, reversibility, and degree of coupling in energy conservation by the mitochondrial respiratory chain. Commun Biol. 2020;3:451.
104. Dudek J, Hartmann M, Rehling P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim Biophys Acta Mol Basis Dis. 2019;1865:810-21.
105. Popoiu T, Maack C, Bertero E. Mitochondrial calcium signaling and redox homeostasis in cardiac health and disease. Front Mol Med. 2023;3:1235188.
106. Davies KM, Strauss M, Daum B, et al. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci U S A. 2011;108:14121-6.
107. Strauss M, Hofhaus G, Schröder RR, Kühlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27:1154-60.
108. Quintana-cabrera R, Quirin C, Glytsou C, et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat Commun. 2018;9:3399.
110. Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005;280:29403-8.
111. Lesnefsky EJ, Hoppel CL. Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochim Biophys Acta Bioenerg. 2008;1777:1020-7.
112. Du J, Yu Q, Anjorin OE, Wang M. Age-related mitochondrial alterations contribute to myocardial responses during sepsis. Cells. 2025;14:1221.
113. Gherardi G, Monticelli H, Rizzuto R, Mammucari C. The mitochondrial Ca2+ uptake and the fine-tuning of aerobic metabolism. Front Physiol. 2020;11:554904.
114. Mather M, Rottenberg H. Aging enhances the activation of the permeability transition pore in mitochondria. Biochem Biophys Res Commun. 2000;273:603-8.
115. Bertero E, Nickel A, Kohlhaas M, et al. Loss of mitochondrial Ca2+ uniporter limits inotropic reserve and provides trigger and substrate for arrhythmias in Barth syndrome cardiomyopathy. Circulation. 2021;144:1694-713.
116. Yan X, Wang R, Xu H, Tao Z, Ling J. The mechanisms associated with inflammation and coronary microvascular dysfunction in heart failure with preserved ejection fraction. Med Princ Pract. 2025;35:101-13.
117. Patyal P, Azhar G, Verma A, et al. Mitochondrial dynamics in aging heart. Biomedicines. 2025;13:2603.
118. Vásquez‐trincado C, García‐carvajal I, Pennanen C, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 2016;594:509-25.
119. Nakayama H, Otsu K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem J. 2018;475:839-52.
120. Guerrero-navarro L, Jansen-dürr P, Cavinato M. Age-related lysosomal dysfunctions. Cells. 2022;11:1977.
121. Rodríguez-nuevo A, Zorzano A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress. 2019;3:195-207.
122. Grazioli S, Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front Immunol. 2018;9:832.
123. Chen S, Liao Z, Xu P. Mitochondrial control of innate immune responses. Front Immunol. 2023;14:1166214.
124. Zhang B, Chang JY, Lee MH, Ju S, Yi H, Shong M. Mitochondrial stress and mitokines: therapeutic perspectives for the treatment of metabolic diseases. Diabetes Metab J. 2024;48:1-18.
125. Konerman MC, Greenberg JC, Kolias TJ, et al. Reduced myocardial flow reserve is associated with diastolic dysfunction and decreased left atrial strain in patients with normal ejection fraction and epicardial perfusion. J Card Fail. 2018;24:90-100.
126. Janaszak-jasiecka A, Płoska A, Wierońska JM, Dobrucki LW, Kalinowski L. Endothelial dysfunction due to eNOS uncoupling: molecular mechanisms as potential therapeutic targets. Cell Mol Biol Lett. 2023;28:21.
127. Lipowsky HH. The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Ann Biomed Eng. 2011;40:840-8.
128. Lagrange J, Jahangiri M, Baudry G, et al. Association between endothelial alterations, cardiac function, and outcomes from health to heart failure: insight from the STANISLAS, MEDIA‐DHF, and BIOSTAT‐CHF cohorts. J Am Hear Assoc. 2025;14:e040179.
129. Aldiwani H, Nelson MD, Sharif B, et al. Reduced myocardial perfusion is common among subjects with ischemia and no obstructive coronary artery disease and heart failure with preserved ejection fraction: a report from the WISE-CVD continuation study. Vessel Plus. 2022;6:16.
130. Su H, Cantrell AC, Zeng H, Zhu S, Chen J. Emerging role of pericytes and their secretome in the heart. Cells. 2021;10:548.
131. Armulik A, Genové G, Mäe M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557-61.
132. O'farrell FM, Mastitskaya S, Hammond-haley M, Freitas F, Wah WR, Attwell D. Capillary pericytes mediate coronary no-reflow after myocardial ischaemia. eLife. 2017;6:e29280.
133. Simmonds SJ, Grootaert MOJ, Cuijpers I, et al. Pericyte loss initiates microvascular dysfunction in the development of diastolic dysfunction. Eur Heart J Open. 2024;4:oead129.
134. Das A, Huang GX, Bonkowski MS, et al. Impairment of an endothelial NAD+-H2S signaling network is a reversible cause of vascular aging. Cell. 2018;173:74-89.e20.
135. Grossini E, Venkatesan S, Ola Pour MM. Mitochondrial dysfunction in endothelial cells: a key driver of organ disorders and aging. Antioxidants. 2025;14:372.
136. Han Q, Yu Y, Liu X, et al. The Role of endothelial cell mitophagy in age-related cardiovascular diseases. Aging Dis. 2025;16:2151.
137. Csiszar A, Tarantini S, Yabluchanskiy A, et al. Role of endothelial NAD+ deficiency in age-related vascular dysfunction. Am J Physiol Heart Circ Physiol. 2019;316:H1253-66.
138. Sinha A, Rahman H, Webb A, Shah AM, Perera D. Untangling the pathophysiologic link between coronary microvascular dysfunction and heart failure with preserved ejection fraction. Eur Heart J. 2021;42:4431-41.
139. Huang LS, Hong Z, Wu W, et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. 2020;52:475-486.e5.
140. Singh M, Jhajharia A, Pruthi R, Carmichael OT. 31P‐MRS‐measured phosphocreatine recovery kinetics in human muscles in health and disease-a systematic review and Meta‐analysis. NMR Biomed. 2025;38:e70023.
141. Georgakoudi I, Quinn KP. Label-free optical metabolic imaging in cells and tissues. Annu Rev Biomed Eng. 2023;25:413-43.
142. Chacko BK, Kramer PA, Ravi S, et al. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Investig. 2013;93:690-700.
143. Trumpff C, Michelson J, Lagranha CJ, et al. Stress and circulating cell-free mitochondrial DNA: a systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion. 2021;59:225-45.
144. Miotto PM, Mcglory C, Holloway TM, Phillips SM, Holloway GP. Sex differences in mitochondrial respiratory function in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2018;314:R909-15.
145. De Goede P, Wefers J, Brombacher EC, Schrauwen P, Kalsbeek A. Circadian rhythms in mitochondrial respiration. J Mol Endocrinol. 2018;60:R115-30.
146. Liu D, Li H, Lu J, Bai Y. Tissue-specific implications of mitochondrial alterations in aging. Front Biosci. 2013;E5:734-47.
147. Thijssen DHJ, Bruno RM, Van Mil ACCM, et al. Expert consensus and evidence-based recommendations for the assessment of flow-mediated dilation in humans. Eur Heart J. 2019;40:2534-47.
148. Pence BD, Yarbro JR. Aging impairs mitochondrial respiratory capacity in classical monocytes. Exp Gerontol. 2018;108:112-7.
149. Georgakoudi I, Skala MC, Quinn KP, et al. Consensus guidelines for cellular label-free optical metabolic imaging: ensuring accuracy and reproducibility in metabolic profiling. J Biomed Opt. 2025;30:S23901-S23901.
150. Pfeiffer K, Gohil V, Stuart RA, et al. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873-80.
151. Panel M, Ghaleh B, Morin D. Mitochondria and aging: a role for the mitochondrial transition pore? Aging Cell. 2018;17:e12793.
152. Sindler AL, Delp MD, Reyes R, Wu G, Muller‐delp JM. Effects of ageing and exercise training on eNOS uncoupling in skeletal muscle resistance arterioles. J Physiol. 2009;587:3885-97.
153. Liang W, Sagar S, Ravindran R, et al. Mitochondria are secreted in extracellular vesicles when lysosomal function is impaired. Nat Commun. 2023;14:5031.
154. Kim J, Kim H, Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. 2023;55:510-9.
155. Seals D, Murphy M, Gioscia-Ryan R, et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension. 2018;71:1056-63.
157. Dorn GW. Mitochondrial pruning by nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010;3:374-83.
158. Titus AS, Sung E, Zablocki D, Sadoshima J. Mitophagy for cardioprotection. Basic Res Cardiol. 2023;118:42.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.