


Cardiac remodeling and healthy aging: mechanisms and adaptive functional changes
 0
0 Abstract
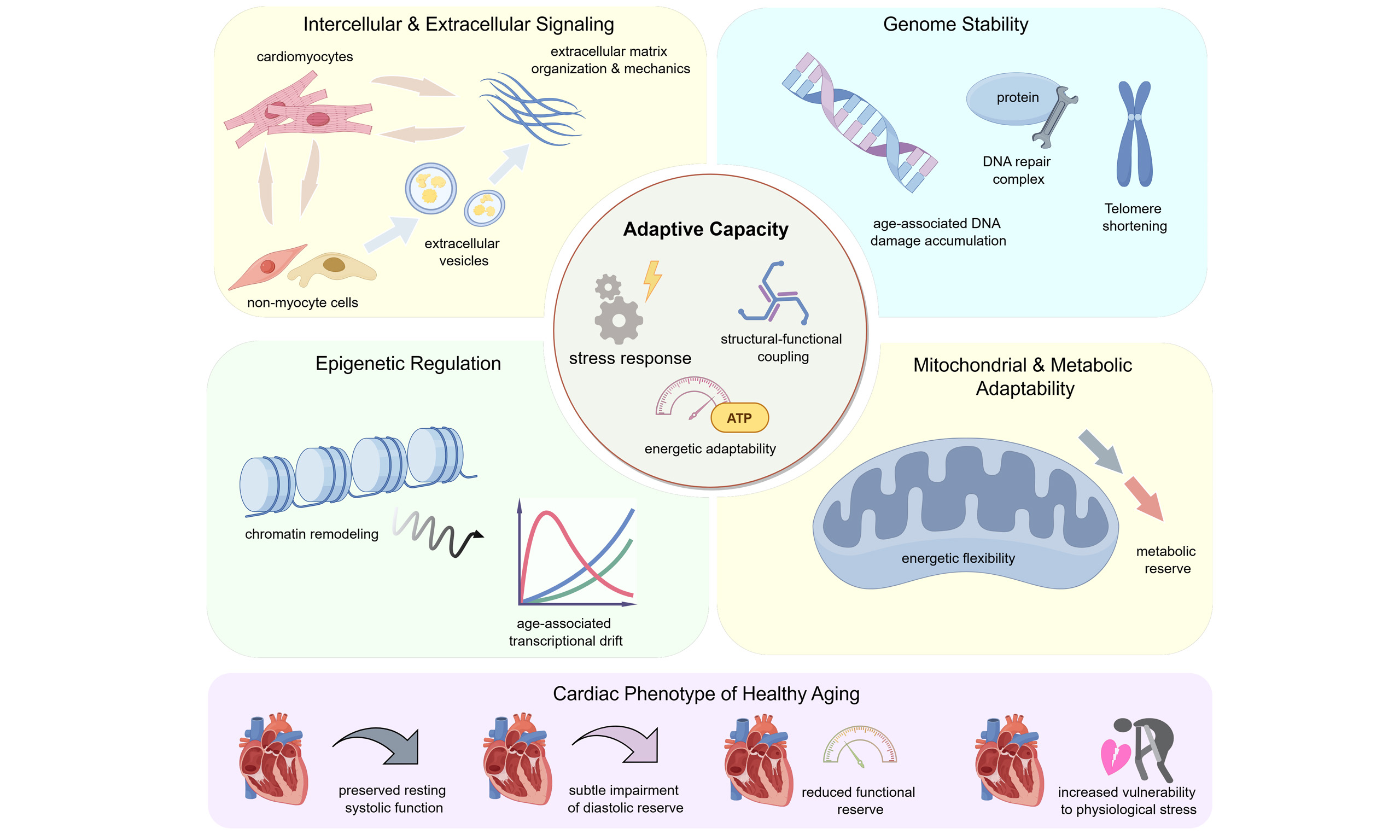
Cardiac aging is accompanied by progressive alterations in myocardial structure and function that arise even in the absence of overt cardiovascular disease. Large population-based imaging studies demonstrate that healthy aging is typically characterized by concentric geometric remodeling, subtle impairment of diastolic performance, and preserved resting systolic function, alongside a gradual decline in cardiac functional reserve. These features indicate that age-related cardiac remodeling does not simply reflect pathological degeneration, but instead encompasses a range of adaptive responses with substantial inter-individual variability. Mechanistically, remodeling of the aging heart reflects coordinated changes across multiple regulatory layers. Alterations in intercellular communication, extracellular matrix organization, mitochondrial quality control, DNA damage responses, and epigenetic regulation collectively influence myocardial stiffness, energetic flexibility, and responsiveness to physiological stress. While many of these processes may initially support cardiac performance, their progressive dysregulation can reduce compensatory capacity and increase vulnerability to stress with advancing age. This review integrates current evidence on the structural, functional, and molecular features of cardiac remodeling during healthy aging, with an emphasis on biological aging mechanisms rather than disease-driven remodeling. We further discuss emerging strategies that prioritize preservation of functional reserve, modulation of extracellular matrix mechanics, and maintenance of mitochondrial and metabolic adaptability. Viewing cardiac aging through the lens of adaptive capacity provides a framework for understanding how physiological remodeling transitions toward functional limitation and for developing interventions aimed at sustaining cardiovascular healthspan rather than reversing established age-related structural changes.
Keywords
INTRODUCTION
The aging process brings about progressive changes in the structure and function of the heart, which collectively influence the health of the human cardiovascular system[1]. Notably, age-related cardiac remodeling occurs even without clinically overt cardiovascular disease, involving coordinated changes in chamber geometry, myocardial compliance, and myocardial metabolism[1,2]. These age-related features are highly prevalent and contribute to the increased susceptibility of older individuals to physiological and pathological stress[3,4]. However, it remains unclear whether such remodeling primarily reflects inevitable functional decline or instead represents adaptive processes that support cardiac performance during healthy aging. It is crucial to recognize that cardiac aging should be distinguished from disease-driven myocardial remodeling. Pathological remodeling typically arises from defined insults, including hypertension, ischemia, or inherited cardiomyopathies, whereas healthy aging encompasses physiological adaptations occurring in individuals without apparent cardiac disease[5-7]. Population-based and imaging studies indicate that detectable structural changes in the heart may emerge as early as midlife, with trends toward concentric remodeling and reduced chamber volume reported in individuals between 40 and 50 years of age[8]. However, the onset, trajectory, and severity of age-related cardiac remodeling vary markedly among individuals. Comparisons of cardiac function between octogenarians and centenarians further illustrate that advanced chronological age does not uniformly dictate myocardial preservation, as some exceptionally long-lived individuals retain relatively preserved cardiovascular regulation[9]. In this context, chronological age alone provides limited insight into cardiac health. While it offers a convenient temporal framework for population-level analyses, chronological age fails to capture the substantial biological heterogeneity observed in cardiac aging. In contrast, biological aging reflects cumulative functional and molecular alterations that more directly determine myocardial structure, resilience, and susceptibility to stress and disease[10].
Growing evidence indicates that cardiac remodeling during healthy aging is not solely a passive degenerative process[11]. Modest increases in myocardial mass, alterations in diastolic properties, and shifts in substrate preference may reflect compensatory responses to evolving energetic demands, mechanical loading, and systemic metabolic cues[2,12]. At the cellular level, advancing age is associated with coordinated remodeling of mitochondrial function and redox signaling, alongside increasing heterogeneity in intercellular communication within the myocardium[13]. As global life expectancy continues to rise, preserving cardiac function has become a central objective of cardiovascular medicine, rather than focusing exclusively on the treatment of established disease. A clearer understanding of adaptive cardiac responses to aging, and of the thresholds at which these responses fail, may guide efforts to preserve functional reserve, improve stress tolerance, and prolong healthspan. In this review, we integrate current evidence on cardiac remodeling during healthy aging, with a focus on mechanisms and interventions that target biological aging processes rather than disease states.
CARDIAC REMODELING IN HEALTHY AGING
Several large observational cohorts, in which overt cardiovascular disease and major cardiac pathology are rigorously excluded, have used standardized echocardiography and cardiovascular magnetic resonance to systematically examine myocardial remodeling during healthy aging[14,15]. Across cohorts spanning early adulthood to advanced age, aging is consistently associated with a reduction in left ventricular end-diastolic volume, while global systolic function, as assessed by ejection fraction, remains preserved[8,15]. More sensitive indices, particularly those reflecting longitudinal systolic function, suggest mild decline with advancing age even in the absence of overt cardiovascular disease, including lower tissue Doppler-derived systolic velocities, reduced mitral annular plane systolic excursion, and less favorable longitudinal strain patterns[16,17]. This pattern results in an increased left ventricular mass-to-volume ratio, reflecting a shift toward concentric remodeling rather than ventricular dilatation. Importantly, these geometric changes are observed in the absence of myocardial infarction, significant valvular disease, or pathological ventricular enlargement, supporting their interpretation as intrinsic features of cardiac aging rather than unrecognized disease processes.
Beyond chamber geometry, functional imaging reveals that age-related remodeling is accompanied by subtle but measurable alterations in diastolic performance. Doppler and strain-based analyses demonstrate slower myocardial relaxation, reduced early diastolic filling, and lower peak diastolic strain rates with advancing age, despite preserved systolic indices[8,15]. These changes develop gradually from midlife and progress over decades, indicating a slow remodeling trajectory rather than abrupt structural deterioration. Tissue characterization studies further refine the distinction between healthy aging and pathological remodeling. Native T1 mapping and related techniques suggest modest age-related changes in myocardial composition, which differ in magnitude and pattern from those observed in hypertensive heart disease or cardiomyopathies[14,18]. Together, these imaging-based observations establish cardiac remodeling as a common and quantifiable feature of healthy aging, characterized by preserved pump function, concentric geometric adaptation, and heterogeneous trajectories across individuals.
MECHANISMS OF CARDIAC REMODELING IN HEALTHY AGING
Cardiac aging involves progressive alterations in multiple communication levels, ranging from extracellular signaling to intracellular dysfunction, which collectively contribute to structural and functional remodeling of the heart.
Cellular communication mechanisms
Dysregulation of paracrine/endocrine signaling
Aging accelerates the senescence of surrounding cells and organs through paracrine mechanisms[19]. Hu et al. combined a D-galactose induced rat model with cell-based experiments and showed that mesenchymal stem cell treatment attenuated aging-related cardiac injury, accompanied by increased SOD2 expression and reduced oxidative stress and p53 signaling. Their conditioned medium experiments suggested that insulin-like growth factor 1 (IGF-1) may be one of the paracrine mediators involved, although direct in vivo validation of this specific mechanism remains limited[20]. Senescent cells secrete a large number of senescence-associated secretory phenotype (SASP) factors, including cytokines [such as Interleukin (IL)-1α, IL-1β, and IL-6], chemokines (such as IL-8 and growth-regulated-α protein), growth factors (such as fibroblast growth factor 2 and hepatocyte growth factor), and matrix metalloproteinases [such as Matrix Metallopeptidase 1 (MMP1), MMP3, and MMP13]. These secreted mediators act on neighboring cells in a paracrine manner, triggering chronic inflammatory responses, disrupting the tissue microenvironment, and accelerating the aging process[21].
The endocrine system regulates the functions of multiple systems within the body by using signaling molecules such as hormones, which act on distant target cells via the bloodstream. During the aging process, endocrine function gradually declines, leading to changes in hormone levels. In addition to conventional endocrine signals, parabiosis-based studies have suggested that age-related cardiac phenotypes can also be shaped by circulating systemic factors. A seminal study by Loffredo et al. showed that exposure to a young circulation attenuated age-related cardiac hypertrophy in old mice and proposed Growth Differentiation Factor 11 (GDF11) as a candidate rejuvenating factor[22]. However, later studies reported conflicting findings regarding age-related changes in GDF11 and its functional effects[23,24]. Thus, while parabiosis-based work strongly supports the concept that blood-borne factors influence cardiac aging, the specific contribution of GDF11 remains controversial. Beyond these circulating factors, age-related changes in classical endocrine mediators have also been implicated in cardiac aging. Research has confirmed that aging is associated with a significant decrease in androgen levels[25]. Furthermore, estrogen replacement therapy has been shown to effectively slow down cardiac aging[26]. Some studies also indicate that sex, sex hormones, and sex chromosomes are key factors influencing cardiac aging[27]. The renin-angiotensin system is another important hormonal regulator of cardiovascular function during aging. Klotho, which either functions as an obligatory co-receptor for fibroblast growth factor 23 (FGF23) or as a soluble pleiotropic endocrine hormone (s-Klotho), exhibits anti-aging properties[28]. Notably, Klotho can block or inhibit aging-related pathways, such as transforming growth factor beta (TGF-β), IGF-1, nuclear factor kappa B (NF-κB), and Wingless-related integration site (Wnt)/β-catenin, independent of FGF23[28]. Klotho deficiency contributes to cardiac aging by impairing the Nrf2-GR pathway. Supplementation with exogenous secretory Klotho represents a promising therapeutic strategy for treating aging-related cardiomyopathy and heart failure[29]. The protective effects of melatonin on aging-induced alterations in cardiac function and structure offer potential as a new source for rejuvenating the heart[30].
Aging is associated with an increase in epicardial adipose tissue (EAT), which can affect both the function and morphology of pericardial fat[21,31]. Elevated EAT is linked to increased production and secretion of pro-inflammatory mediators and neurohormones. Consequently, thickened EAT may pathologically influence cardiac structure and function through both paracrine and endocrine mechanisms[21]. These alterations in paracrine and endocrine signaling not only affect cellular behavior but also reshape the extracellular microenvironment, thereby contributing to structural remodeling of the aging myocardium.
Extracellular matrix remodeling
Senescent fibroblasts are characterized by alterations in the expression patterns of genes related to inflammation, extracellular matrix, angiogenesis, and osteogenesis[32]. These changes affect the microenvironment and may lead to increased matrix stiffness. Since heightened matrix stiffness can induce telomere shortening-associated contractile dysfunction[33], and altered mechanical stimulation in cardiac fibroblasts (CFs) can result in mechanically induced premature senescence-like phenotypes[34], it is hypothesized that changes in the extracellular environment driven by senescent cardiac fibroblasts may contribute to an accelerated aging phenotype. The aging heart exhibits diastolic dysfunction due to increased collagen deposition. One study found that 5-aminoimidazole-4-carboxamide riboside (AICAR) can downregulate Gli1-dependent collagen expression by activating AMP-activated Protein Kinase (AMPK), thereby reducing fibroblast stimulation and delaying age-related diastolic dysfunction in female mouse hearts. In contrast, the same drug and dosage had minimal effects on fibroblast phenotypes or the Extracellular Matrix (ECM) in aging male mouse hearts, with no specific mitigation of age-related changes in diastolic function[35]. This suggests that extracellular matrix remodeling during cardiac aging may be regulated in a sex-dependent manner[35]. Sex-specific differences may also influence fibrotic signaling, fibroblast activation, and myocardial stiffness during aging, indicating that mechanistic pathways identified in one sex may not fully translate to the other[35,36]. This issue should therefore be taken into account when interpreting mechanistic studies and candidate interventions in cardiac aging. Fibulin 7 (FBLN7) knockout attenuates age-related myocardial fibrosis by promoting TGFBR3/ALK1/Smad1 signaling and inhibiting the pro-fibrotic phenotype of cardiac fibroblasts[37]. Importantly, extracellular matrix remodeling is closely linked to changes in pericyte function. As matrix composition and stiffness evolve during aging, they may influence the behavior of perivascular cells such as pericytes, which play a critical role in maintaining myocardial integrity. These ECM-associated changes are closely linked to subsequent alterations in cell-cell communication within the aging cardiac microenvironment.
ECM-associated intercellular interactions
In the aging heart, intercellular communication extends beyond matrix remodeling and involves dynamic crosstalk among cardiomyocytes, macrophages, endothelial cells, and perivascular cells[38]. Studies indicate that these interactions are required not only for injury responses but also for the maintenance of myocardial homeostasis during aging. Nicolás-Ávila et al. found that resident cardiac macrophages have been shown to remove dysfunctional mitochondria released by cardiomyocytes, thereby helping preserve mitochondrial quality control and cardiac function[39,40]. Endothelial cells regulate cardiomyocyte behavior through paracrine and metabolic signals that coordinate myocardial perfusion, growth, and stress adaptation. Age-related disruption of endothelial-cardiomyocyte crosstalk may therefore contribute to impaired vascular support and functional decline[41,42]. In addition, changes in pericyte function also participate in this intercellular remodeling. Research findings indicate that RGS5 is a key regulator of pericyte function during cardiac aging. Loss of RGS5 leads to cardiac dysfunction and induces myocardial fibrosis, a hallmark of cardiac aging[43]. These intercellular communication changes provide a basis for the emergence of non-classical signaling pathways in the aging heart.
Together, these changes indicate that cardiac aging is shaped by coordinated alterations in paracrine and endocrine signaling, extracellular matrix remodeling, and intercellular interactions [Figure 1].
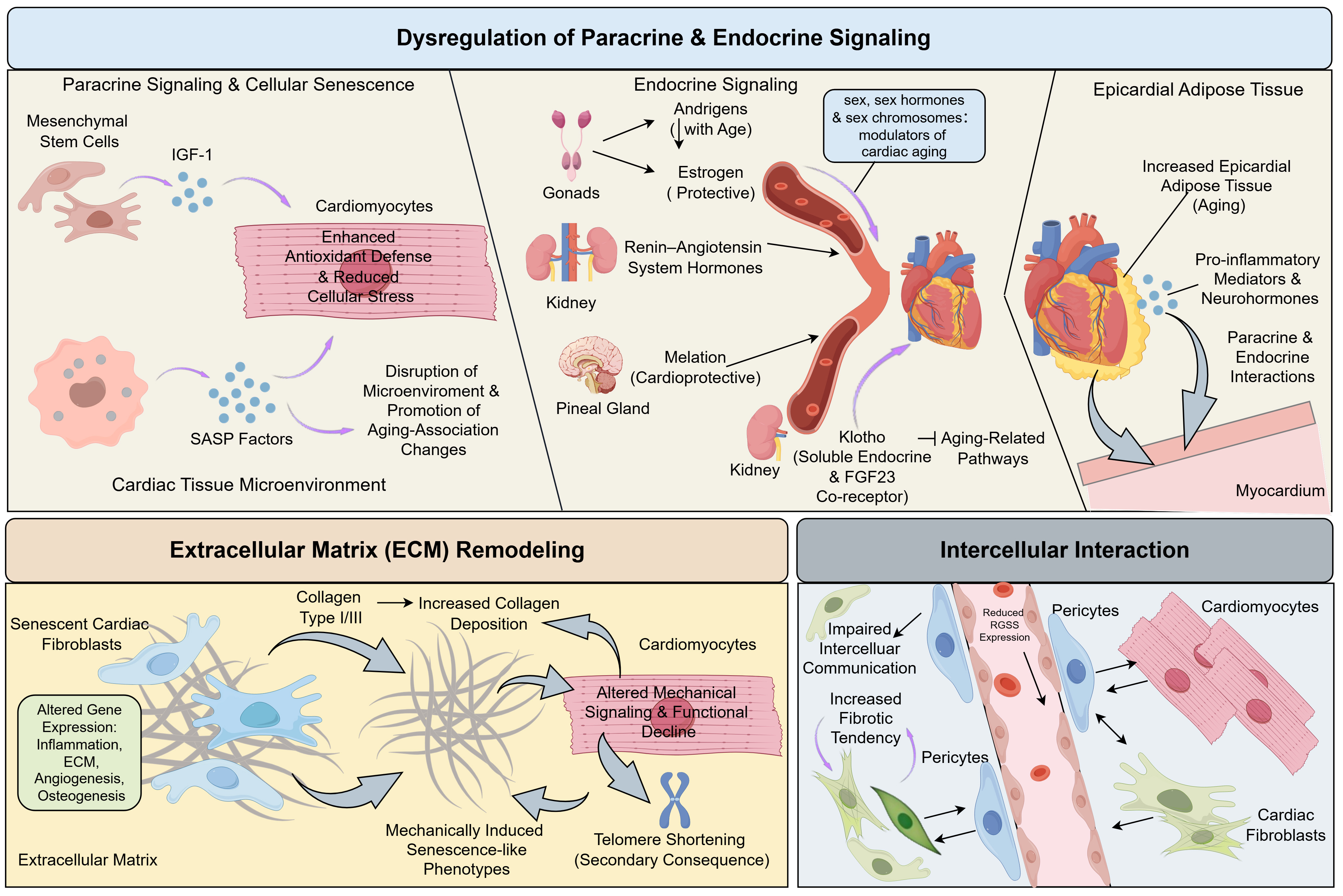
Figure 1. Dysregulation of Paracrine and Endocrine Signaling, extracellular matrix (ECM) Remodeling, and Intercellular Interactions. Paracrine and endocrine signaling, along with changes in the ECM and intercellular interactions, contribute to cardiac aging. In youthful hearts, mesenchymal stem cells (MSCs) release insulin-like growth factor-1 (IGF-1), which enhances SOD2 expression, reducing oxidative stress. However, with aging, senescent cells secrete factors such as cytokines, chemokines, and growth factors, disrupting the tissue microenvironment and accelerating aging. Klotho, a pleiotropic hormone, helps mitigate these aging processes by modulating pathways like TGF-β and NF-κB. In aged hearts, senescent fibroblasts contribute to increased matrix stiffness and diastolic dysfunction through altered collagen deposition. Additionally, loss of RGS5 in pericytes leads to myocardial fibrosis, a hallmark of aging in the heart. The accumulation of epicardial adipose tissue (EAT) also promotes the secretion of pro-inflammatory mediators, further exacerbating cardiac aging. Created by figdraw.com. (Copyright Code: PYYST71664). SASP: Senescence-associated secretory phenotype; TGF-β: transforming growth factor beta; NF-κB: nuclear factor kappa B.
Alterations in non-classical communication pathways
Extracellular vesicle mediated communication
Extracellular vesicles (EVs), as critical mediators of intercellular communication, are released by various cell types and possess a lipid bilayer membrane structure. They can encapsulate and transport a variety of bioactive molecules, playing a significant role in both physiological and pathological processes[44]. Studies have found that a youthful systemic environment can impart rejuvenating characteristics to aged cells or organs. EVs, acting as carriers of proteins and genetic material, play a key role in mediating the beneficial effects of a young systemic environment on aging[45]. Chen et al. study has shown that small extracellular vesicles derived from young mouse plasma can reverse multiple age-associated phenotypes in aged mice, including improvements in mitochondrial function and systemic metabolic capacity, largely through miRNA-mediated regulation of energy metabolism pathways[46]. Similarly, EVs isolated from young serum have been reported to attenuate cellular senescence and restore mitochondrial and immunometabolic function in aged tissues[47]. It is hypothesized that EVs may carry biologically active molecules with anti-aging properties, regulating intracellular signaling pathways to reverse the senescent cell phenotype. Small extracellular vesicles (sEVs) derived from young adipose-derived stem cells represent a novel, non-invasive therapeutic strategy with the potential to mitigate aging-related functional decline in the heart[48]. Treatment with young cardiosphere-derived cell (CDC) EVs improves both the structure and function of the heart in aged rats, while also favorably modulating glucose metabolism and anti-aging pathways[45]. Overall, EVs may serve as endogenous regulators of aging.
Metabolite signaling
In the field of metabolite signaling, multiple studies have revealed the significant impact of specific metabolites on cardiovascular health and the aging process. Long-term dietary intervention with alpha-linolenic acid downregulates the expression of pro-inflammatory and pro-oxidative regulators, inhibits markers of ECM remodeling, and thereby prevents late-stage cardiac fibrosis and arteriosclerosis in mouse models of chronic aging[49]. The nicotinamide adenine dinucleotide (NAD) pool tends to decline with normal aging, obesity, and hypertension, which are key risk factors for cardiovascular diseases. NAD supplementation has been shown to extend healthspan, prevent metabolic syndrome, and lower blood pressure in preclinical models[50]. Taurine, known for its cardioprotective effects, decreases with age. Counteracting this decline through taurine supplementation may delay the development of age-related health issues[51].
Together, these findings indicate that non-classical communication pathways participate in cardiac aging. By influencing mitochondrial function, energy metabolism, inflammatory signaling, and extracellular matrix remodeling, these systemic signals can modulate cardiomyocyte homeostasis and thereby contribute to age-related structural and functional alterations in the heart [Figure 2].
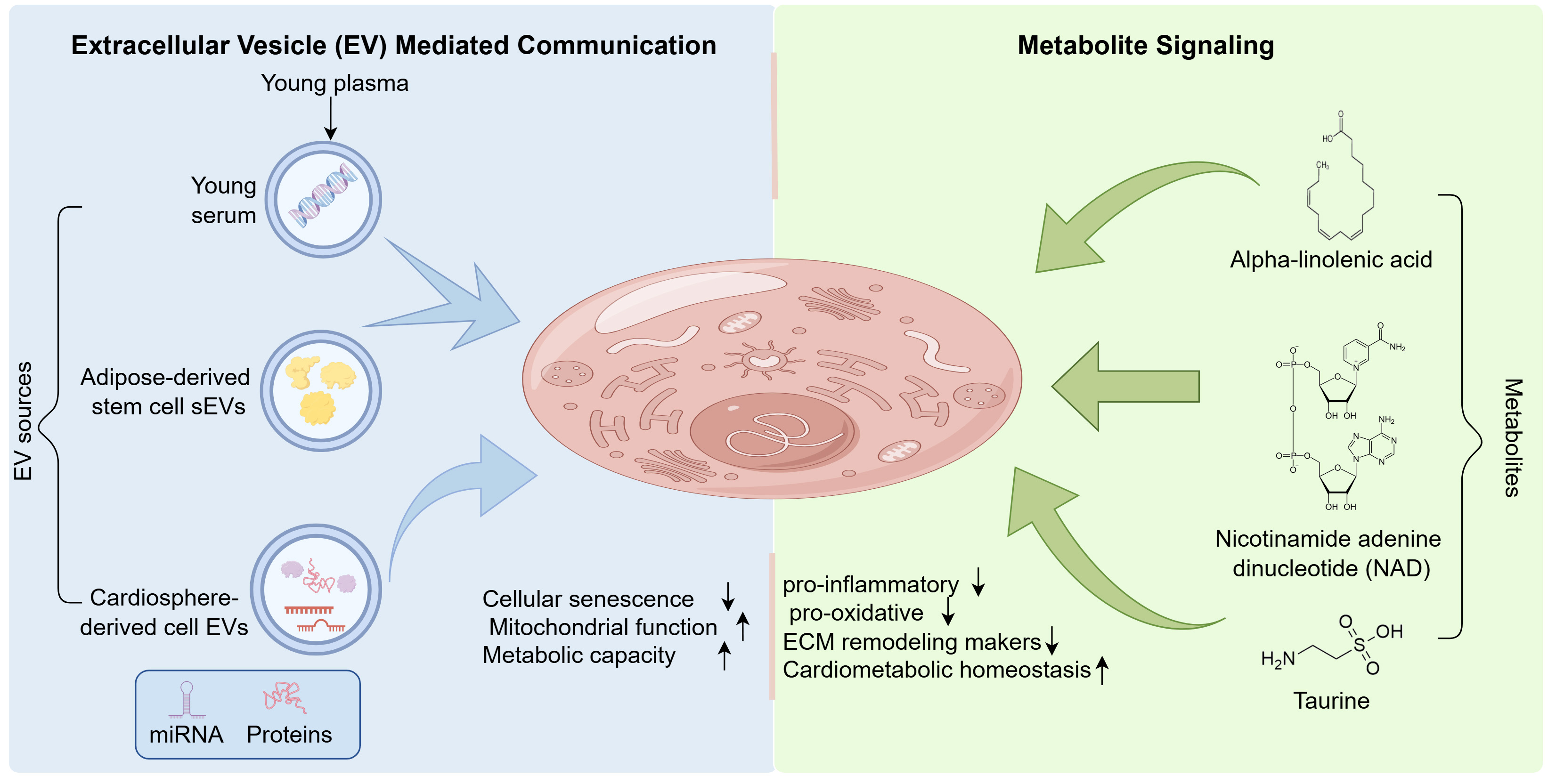
Figure 2. Alterations in non-classical communication pathways. Extracellular vesicles released from young plasma, young serum, adipose-derived stem cells, and cardiosphere-derived cells are shown to deliver miRNAs and proteins to recipient cardiomyocytes. Such vesicle-mediated transfer is associated with reduced cellular senescence and improved mitochondrial function and metabolic capacity. In parallel, metabolite signaling is represented by alpha-linolenic acid, nicotinamide adenine dinucleotide, and taurine, which are associated with reduced inflammatory and oxidative stress signaling, reduced extracellular matrix remodeling markers, and maintenance of cardiometabolic homeostasis. Created by figdraw.com. (Copyright Code: UYISR4d900). ECM: Extracellular matrix; sEVs: small extracellular vesicles.
Intracellular mechanisms of functional decline
Imbalance between DNA damage and repair
In the process of cardiac aging, the accumulation of DNA damage and the decline in repair mechanisms constitute core pathological links. Aging myocardial tissues commonly exhibit phenomena such as DNA single-strand breaks, double-strand breaks, and oxidative base damage, primarily stemming from the continuous generation of endogenous reactive oxygen species (ROS) under the high-metabolic state of cardiomyocytes[52,53]. Oxidative stress and metabolic disorders, which intensify with age, further amplify this damage burden.
Excessive DNA damage directly induces cardiomyocyte senescence, characterized by the upregulation of cell cycle inhibitors such as p16INK4a and p21, and activation of the SASP, leading to the release of a large number of inflammatory cytokines. This not only impairs the function of cardiomyocytes themselves but also promotes cardiac fibrosis and remodeling through paracrine effects, accelerating the overall decline in cardiac function[54,55]. Notably, DNA repair capacity in the aging heart shows a significant decline. The expression of key repair genes, such as Excision Repair Cross-Complementing Group 1 (ERCC1) and Xeroderma Pigmentosum Group G (XPG), decreases, resulting in reduced repair efficiency and the inability to promptly clear damage[56]. Studies have demonstrated that specific knockout of DNA repair enzymes in cardiomyocytes can lead to decreased cardiac function, increased fibrosis, and elevated cell apoptosis, directly confirming the driving role of repair defects in cardiac aging. At the molecular pathway level, important repair mechanisms such as non-homologous end joining (NHEJ) are impaired during aging. For example, decreased XRCC4 function activates the p53 signaling pathway, thereby inducing cardiomyocyte apoptosis[57]. Furthermore, the loss of the telomere-binding protein Rap1 has also been shown to exacerbate DNA damage and disrupt the Tumor protein p53/Peroxisome Proliferator-Activated Receptor Alpha (p53/PPARα) signaling axis, jointly promoting the cardiac aging process[58].
In the biological progression of cardiac aging, the functional decline of the telomere system constitutes a key molecular mechanism. Telomeres, as protective structures at the ends of chromosomes, not only mark the exhaustion of cellular replicative potential through their progressive shortening and loss of stability but also directly drive cardiomyocyte senescence by activating DNA damage response pathways. Studies have shown that even in cardiomyocytes with limited proliferative capacity, telomere dysfunction can induce the formation of telomere dysfunction-induced DNA damage foci, activating senescence pathways such as p21CIP and p16INK4a, promoting the formation of a senescent phenotype in cardiomyocytes, and subsequently leading to myocardial hypertrophy, fibrosis, and functional decline[59-61]. Clinical evidence further supports the association between telomere damage and cardiac aging. Histological analysis of patients with chronic heart failure reveals significant telomere shortening in both cardiomyocytes and peripheral blood leukocytes, with telomere length showing an independent negative correlation with cardiac function indicators such as ventricular ejection fraction[62]. Additionally, age-related mitochondrial dysfunction leads to excessive ROS generation, which can directly cause oxidative damage to telomeric DNA, exacerbating telomere instability and loss, forming a telomere-mitochondria vicious cycle that jointly drives the cardiac aging process[61].
Mitochondrial dysfunction
Mitochondrial quality control mechanisms constitute a core defense system for maintaining cardiomyocyte homeostasis and preventing the accumulation of mitochondrial dysfunction. This system operates through multi-level, well-ordered regulatory pathways to preserve integrity from the molecular to the organellar scale. At the molecular level, the antioxidant system serves as the first line of defense by neutralizing reactive oxygen species to limit oxidative damage to mitochondrial components. Once damage occurs, a secondary layer of repair mechanisms is activated, including mitochondrial DNA repair, reductase systems, and chaperone-mediated protein refolding. If damage is irreversible, intra-mitochondrial proteolytic systems degrade abnormal proteins, enabling functional replacement. When damage exceeds the capacity of molecular-level repair, quality control escalates to the organellar level. Moderately damaged mitochondria can undergo fusion with healthy organelles to dilute localized defects, whereas severely damaged ones are segregated through fission and subsequently eliminated via selective mitophagy[63]. Dysfunction at any level of this hierarchical network can lead to cumulative damage, energy metabolism disruption, and ultimately, cellular functional decline. The following sections will systematically elaborate on the regulatory roles and pathological significance of core mitochondrial quality control mechanisms, including mitochondrial DNA homeostasis, bioenergetic dysfunction, and autophagy, in the context of cardiac aging.
Mitochondrial DNA damage and repair mechanisms
Compared with nuclear DNA, mitochondrial DNA is highly susceptible to oxidative damage due to its lack of histone protection, limited repair capacity, and proximity to the electron transport chain. During aging, the efficiency of mitochondrial base excision repair declines, characterized by reduced activity of key enzymes such as 8-oxoguanine glycosylase 1, along with diminished fidelity of DNA polymerase gamma. These changes collectively promote the accumulation of mtDNA damage and mutations, particularly in regions encoding respiratory chain components[64,65]. mtDNA damage impairs cardiomyocyte function through multiple mechanisms, starting with the compromised synthesis of the 13 respiratory chain subunits encoded by mtDNA, which directly disrupts the integrity of the electron transport chain. This results in decreased oxidative phosphorylation efficiency and insufficient Adenosine Triphosphate (ATP) production, ultimately impairing myocardial contractility[66-68]. mtDNA damage further erodes mitochondrial membrane potential, predisposing the permeability-transition pore to open and release cytochrome c, events that launch the apoptotic program. Cytosolic leakage of mtDNA fragments then ignites innate immunity via the cGAS-STING axis, fuelling chronic inflammation that reciprocally amplifies oxidative stress and propels interstitial fibrosis and maladaptive remodeling[69-71].
Mitochondrial bioenergetic dysfunction
During cardiac aging, the decline in electron transport chain (ETC) function has been widely documented, yet the extent of this decline, its specificity across different cardiac cell types, and its causal relationship with myocardial energy transfer systems remain subjects of ongoing debate. Although many studies report reduced activity of ETC complexes I-IV with advancing age, other evidence indicates that deterioration of the creatine kinase (CK) system, the principal catalyst of rapid intracellular energy transfer and buffering, may correlate more closely with impaired myocardial contractility[72,73]. These findings suggest that bioenergetic dysfunction in the aging heart is unlikely to arise from a single dominant lesion. Instead, impaired oxidative phosphorylation, defective phosphocreatine-mediated energy transfer, and reduced energetic reserve may act together to limit myocardial performance[73,74]. Specifically, energy metabolism disorder in the aging heart has often been described in terms of a global decline in ETC function. Beyond the reduced activity of respiratory chain complexes, impaired function of the CK system further exacerbates energy supply insufficiency. This enzyme system plays a central role in maintaining the dynamic balance between phosphocreatine and ATP, and its decreased activity weakens intracellular energy buffering capacity, directly compromising the immediate energy supply during myocardial contraction[75]. Taken together, current evidence supports the view that energy metabolism impairment in cardiac aging reflects coordinated abnormalities in ATP production, energy transfer, and energy buffering, rather than an isolated defect in any single pathway.
Thus, energy metabolism impairment in cardiac aging results from the combined effects of multiple mechanisms, including both diminished ATP production and reduced efficiency of energy storage and turnover[76]. Concurrently, dysregulated NAD+ metabolism represents another critical node in energy metabolic control. NAD+ levels decline by approximately 30%-50% in the aging heart, accompanied by a significant decrease in the NAD+/NADH ratio. This metabolic shift directly impairs the activity of the sirtuin family, with notable suppression of Sirt1 and Sirt6 functions[77,78]. Reduced sirtuin activity subsequently leads to altered histone acetylation states, compromised autophagy, and downregulation of genes involved in mitochondrial biogenesis, thereby establishing a multi-layered dysfunction that spans metabolic disturbance, epigenetic dysregulation, and impaired cellular quality control[79]. Mitochondrial dynamics regulation exhibits a complex pattern of change during aging. Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha (PGC-1α), a master regulator of mitochondrial biogenesis, may show compensatory upregulation in early aging stages, yet its signaling efficiency is diminished. Specifically, although PGC-1α can translocate into the nucleus, its ability to interact with downstream transcription factors such as NRF1 and NRF2 is weakened, resulting in insufficient activation of key factors for mitochondrial DNA replication and transcription, including Transcription Factor A, Mitochondrial (TFAM)[80]. Consequently, mitochondrial biogenesis fails to adequately compensate for functional decline, further aggravating the energetic deficit in the aging heart.
Autophagy dysfunction
During cardiac aging, mitophagic function progressively declines, representing a key mechanism underlying the disruption of cardiomyocyte homeostasis. With advancing age, overall autophagic activity in the heart decreases, characterized by reduced levels of key autophagy-related proteins such as Beclin-1, a lower LC3-II/I ratio, and diminished expression of upstream regulators like Forkhead Box O1 (FOXO1) and Transcription Factor EB (TFEB)[81,82]. This decline in autophagy directly impairs the clearance of damaged mitochondria, leading to the accumulation of dysfunctional organelles, which in turn exacerbates oxidative stress, disrupts energy metabolism, and promotes cellular injury. Studies have demonstrated that enhancing autophagy can effectively mitigate age-related cardiac phenotypes. In genetic models, mice with cardiomyocyte-specific overexpression of the autophagy-related gene Atg5 or those expressing the Becn1F121A mutant exhibit elevated autophagic activity, accompanied by reduced cardiac fibrosis, improved cardiac function, and extended lifespan[83]. Conversely, suppression of autophagy accelerates cardiac aging, promoting myocardial hypertrophy and the accumulation of functionally impaired mitochondria. The regulation of mitophagy is closely linked to the SIRT1-mediated-signaling network. As an NAD+-dependent deacetylase, SIRT1 integrates multiple pathways involved in autophagy, mitochondrial biogenesis, and antioxidant defense. In the aging heart, decreased SIRT1 activity coincides with heightened oxidative damage, while cardiomyocyte-specific deletion of SIRT1 exacerbates cardiac dysfunction, endoplasmic reticulum stress, and apoptosis. These findings suggest that the age-related decline in SIRT1 function may further impair mitophagy by altering the acetylation status of downstream target proteins, thereby establishing a detrimental feedback loop[84]. In summary, the age-related reduction in mitophagic efficiency acts as a significant driver of cardiac aging. The underlying mechanisms involve decreased expression of key autophagy-related molecules and diminished activity of regulatory nodes such as SIRT1, collectively leading to insufficient clearance of damaged mitochondria, aggravated oxidative stress, and destabilization of cardiomyocyte homeostasis. Therapeutic strategies targeting this pathway may offer potential avenues for improving cardiac function in aging.
These intracellular alterations do not occur in isolation. Instead, DNA damage, mitochondrial dysfunction, and impaired autophagy progressively interact and amplify one another, contributing to cumulative cellular injury during cardiac aging [Figure 3]. Collectively, these intracellular alterations represent a major downstream component of cardiac aging and are closely linked to upstream extracellular and systemic changes.
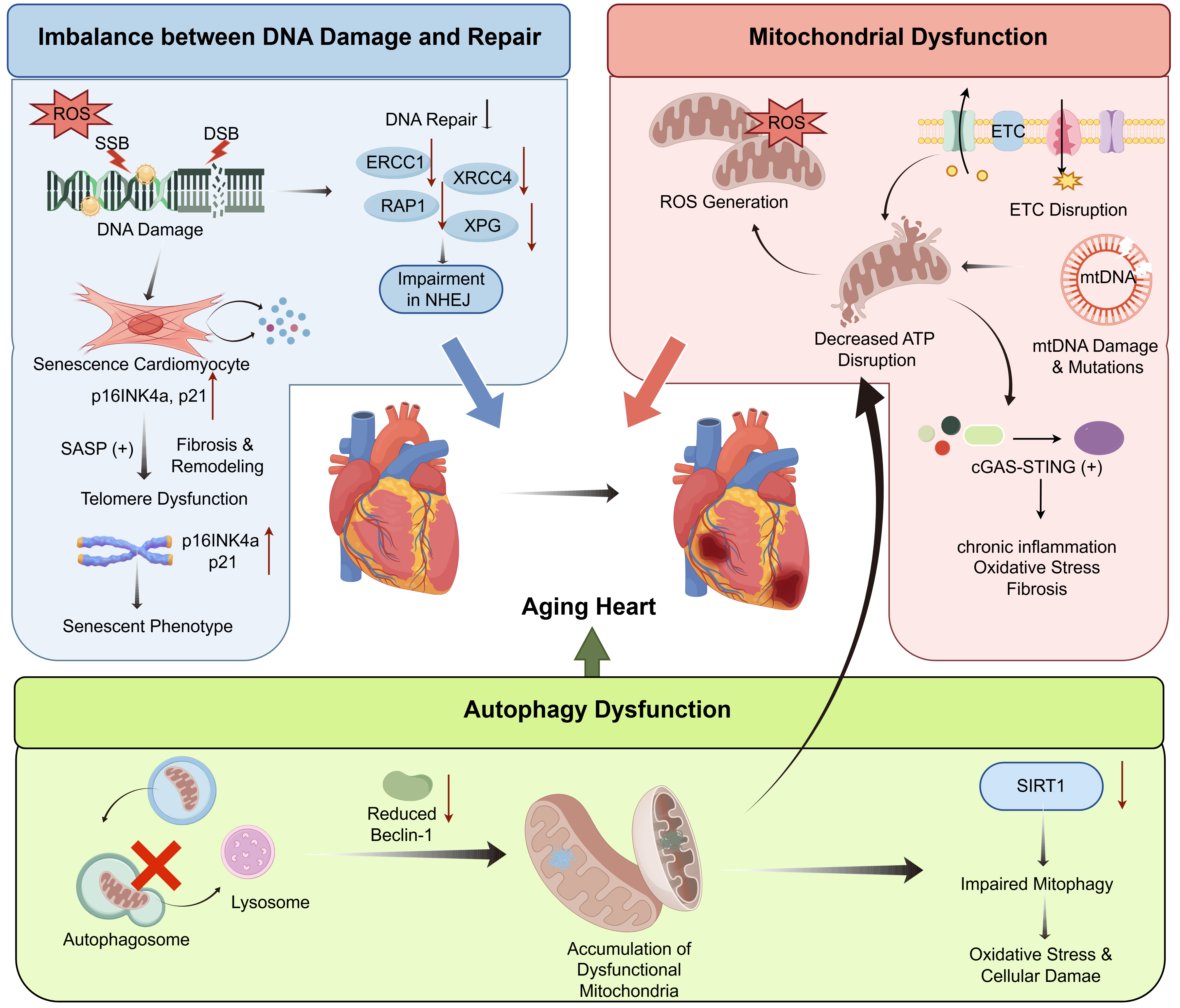
Figure 3. Intracellular mechanisms in cardiac aging. Accumulation of DNA single-strand breaks, double-strand breaks, and oxidative base damage is illustrated in association with elevated reactive oxygen species and impaired DNA repair capacity, including reduced ERCC1, XPG, XRCC4, and disrupted non-homologous end joining. Excessive DNA damage is accompanied by cardiomyocyte senescence marked by increased p16INK4a and p21 expression, activation of senescence-associated secretory phenotype signaling, telomere dysfunction, and fibrotic remodeling. Mitochondrial dysfunction is characterized by enhanced reactive oxygen species generation, disruption of the electron transport chain, mitochondrial DNA damage, and reduced ATP production. Activation of cGAS-STING signaling is shown in association with chronic inflammation, oxidative stress, and fibrosis. Autophagy impairment is depicted by reduced Beclin-1 expression, defective autophagosome–lysosome processing, diminished SIRT1 activity, and accumulation of dysfunctional mitochondria, collectively amplifying oxidative stress and cellular damage. These interconnected intracellular alterations converge on progressive myocardial dysfunction during aging. Created by figdraw.com. (Copyright Code: TIIII906aa). ROS: Reactive oxygen species; SASP: senescence-associated secretory phenotype; NHEJ: non-homologous end joining; ETC: electron transport chain; SSB: Single-strand break; DSB: double-strand break; ATP: adenosine triphosphate; cGAS-STING: cyclic GMP-AMP synthase-stimulator of interferon genes.
Epigenetic dysregulation as a regulatory memory of cardiac aging
Cardiac aging involves a systematic dysregulation of multi-level epigenetic networks, encompassing aberrant DNA methylation patterns, disrupted histone modifications, and dysregulated non-coding RNA expression. These alterations collectively impair transcriptional programs related to oxidative stress, inflammatory responses, angiogenesis, and cellular metabolism, thereby driving progressive deterioration of myocardial structure and function.
Aberrant DNA methylation patterns
At the DNA methylation level, global hypomethylation represents a prominent hallmark of aging, with methylation patterns serving as a biomarker capable of predicting biological age independent of conventional risk factors. Studies indicate that frail individuals exhibit reduced global DNA methylation, and the methylation status of specific genes directly correlates with cardiac functional decline. For instance, hypomethylation of the promoter region of the cell-cycle inhibitor p16INK4a leads to its upregulation, promoting a senescent phenotype in cardiomyocytes. Conversely, hypermethylation of the antioxidant gene GPX1 suppresses its expression, exacerbating oxidative stress injury; selenium supplementation can partially reverse this process by modulating DNMT2 activity. In heart failure, the fibrotic-related gene ITGBL1 shows hypomethylation and increased expression, accelerating cardiac fibrosis progression[85]. Exposure to environmental toxins such as lead and phthalates can also induce sex-specific methylation changes, highlighting the role of environment-epigenetic interactions in cardiac aging. Moreover, DNA methylation-based “epigenetic clocks” have been utilized to assess cardiovascular disease risk and biological age[86,87].
Disordered histone modifications
In the aging heart, increased activity of histone deacetylases results in reduced overall acetylation levels and chromatin compaction, thereby repressing the transcription of cardioprotective genes. Research demonstrates that abnormal accumulation of repressive methylation marks such as H3K27me3 is associated with silencing of fibrosis-related genes, while elevated activity of histone acetyltransferases p300/CBP promotes expression of glycolytic genes and accelerates metabolic reprogramming. Interventions that target these modifying enzymes, such as Histone Deacetylase (HDAC) inhibitors or p300 protein/CREB-binding protein (p300/CBP) antagonists, can improve cardiac function and metabolic state, underscoring the pivotal regulatory role of histone modifications in cardiac aging[88,89].
Dysregulation of non-coding RNA expression
Non-coding RNAs constitute a complex epigenetic regulatory network. Key miRNAs such as miR-34a and miR-21 promote myocardial decline by modulating apoptosis, fibrosis, and inflammatory pathways. The expression of long non-coding RNAs (e.g., the anti-apoptotic lncRNA Sarrah) decreases with age, and its restoration improves cardiac function; some lncRNAs act as competing endogenous RNAs (ceRNAs) that sequester miRNAs and indirectly regulate downstream target genes. Circular RNAs, owing to their stable structure, function as miRNA sponges and participate in the regulation of cardiomyocyte metabolism and proliferation. Together with exosome-mediated intercellular communication, these RNA molecules shape the epigenetic landscape of cardiac aging[90-92]. Lin et al.[93] compared cardiac samples from age-matched controls and patients who suffered sudden cardiac death associated with primary myocardial fibrosis (PMF-SCD). They observed elevated expression of miR-1468-3p in the hearts of elderly PMF-SCD patients relative to healthy elderly individuals. Further mechanistic investigations revealed that miR-1468-3p promotes myocardial fibrosis by enhancing the TGF-β1-Smad3 signaling pathway and drives cardiomyocyte senescence by increasing β-galactosidase activity and the expression of p53 and p16. These findings underscore the dual role of miR-1468-3p in advancing both cardiac fibrosis and cellular aging, offering a potential therapeutic target for age-related cardiac fibrosis[94].
Through effects on senescence, oxidative stress responses, fibrotic signaling, and metabolic regulation, these epigenetic changes contribute to progressive myocardial dysfunction during aging. These epigenetic alterations establish a persistent regulatory memory of aging, reinforcing and stabilizing the functional decline of the aging heart [Figure 4].
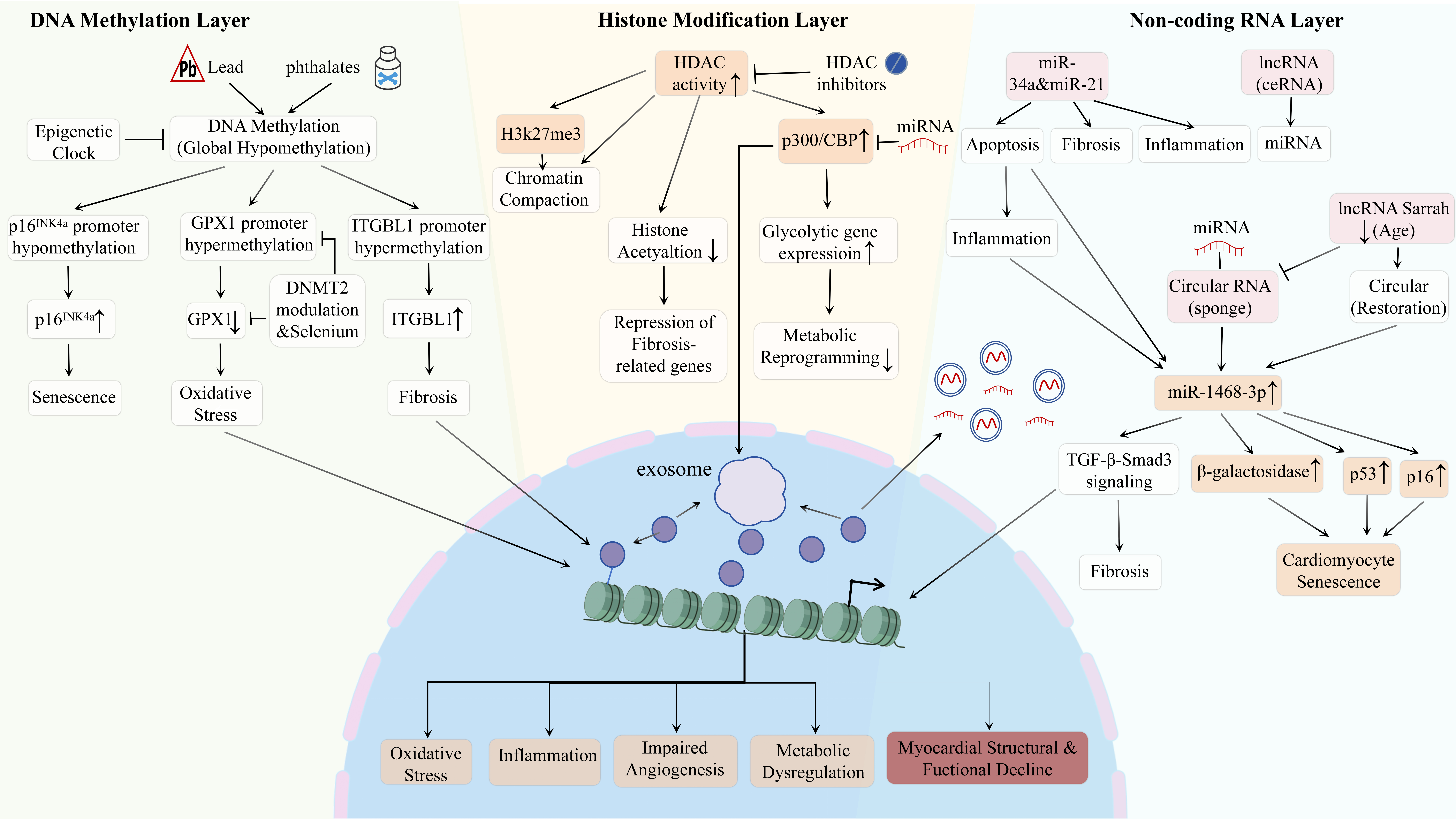
Figure 4. Epigenetic dysregulation in cardiac aging. Aberrant regulation across DNA methylation, histone modification, and non-coding RNA layers is depicted within cardiomyocyte nuclei during aging. Global DNA hypomethylation is associated with epigenetic clock alterations and environmental exposures, accompanied by promoter-specific changes involving p16INK4a, GPX1, and ITGBL1 that link senescence, oxidative stress, and fibrosis. Histone modification imbalance is characterized by increased histone deacetylase activity, reduced acetylation, chromatin compaction, enrichment of H3K27me3, and enhanced p300/CBP-associated metabolic reprogramming. Dysregulated non-coding RNAs, including miR-34a, miR-21, and miR-1468-3p, together with age-related changes in lncRNAs and circular RNAs, modulate apoptotic, fibrotic, and inflammatory pathways through exosome-mediated RNA communication. These coordinated epigenetic alterations converge on transcriptional programs related to oxidative stress, inflammation, angiogenesis, and metabolism, contributing to myocardial structural and functional decline. Created by PowerPoint and figdraw.com. (Copyright Code: TYIUR916b4). TGF-β: Transforming growth factor beta; HDAC: histone deacetylase; CBP: CREB-binding protein.
STRATEGIES AND INTERVENTIONS
In healthy aging, cardiac remodeling reflects the cumulative impact of lifelong mechanical load and metabolic demand rather than discrete pathological insults. Although resting systolic function is frequently preserved, the progressive erosion of functional reserve increases myocardial vulnerability to physiological stress. Accordingly, intervention strategies in healthy aging are not aimed at reversing age-related structural changes, but at delaying maladaptive remodeling and preserving adaptive capacity. The following approaches are grounded in experimental and translational studies conducted in aging models or disease-free populations and focus on modulating biological processes that shape remodeling trajectories rather than restoring youthful cardiac architecture.
Preservation of cardiac functional reserve
In healthy aging, age-related cardiac remodeling is often not accompanied by overt impairment of resting systolic function[14,91,92]. Evidence from longitudinal studies in healthy individuals indicates that reductions in chronotropic responsiveness, impaired diastolic filling during stress, and diminished exercise tolerance often emerge before overt cardiac dysfunction becomes apparent[91,94]. This recognition has prompted increasing attention to strategies that preserve the heart’s capacity to respond to physiological stress rather than focusing only on resting structural parameters. Clinical studies in middle-aged and older adults without overt cardiovascular disease indicate that structured exercise interventions have been shown to improve peak oxygen uptake, chronotropic response, and exercise-related diastolic filling dynamics[95,96]. These functional improvements frequently occur without significant changes in baseline cardiac chamber size or wall thickness, indicating that reserve capacity can be enhanced independently of gross morphological remodeling[96]. Research suggests that interventions targeting physiological responsiveness may mitigate the functional consequences of age-related remodeling, even when structural alterations persist[7,97]. Experimental and clinical studies demonstrate that regular physical activity improves stress-induced cardiac output, enhances diastolic filling under increased demand, and increases peripheral oxygen utilization[98]. These functional improvements are frequently observed in the absence of substantial reversal of age-related structural remodeling.
Caloric restriction and nutrient-sensing pathways
Caloric restriction is one of the extensively studied interventions in the biology of aging and remains highly relevant in the context of cardiac aging[99]. A large of work has linked caloric restriction to delayed functional decline, improved metabolic efficiency, and greater resistance to age-associated stress. These studies have found that these effects converge on AMPK, sirtuins, and Mechanistic Target of Rapamycin (mTOR), which are nutrient-sensing pathways that govern energy balance and cellular maintenance[100-102]. These pathways, in turn, link closely to autophagy, mitochondrial turnover, and stress adaptation in the aging myocardium. Within the cardiovascular system, Caloric Restriction (CR) has been associated with reduced oxidative stress, improved mitochondrial function, and attenuation of age-related metabolic disturbances[103]. Although effect magnitudes and durability vary across experimental models, the collective evidence suggests that CR delays several features of biological aging rather than simply masking them. Fasting-based paradigms and caloric-restriction mimetics have attracted interest because they engage overlapping nutrient-sensing pathways[104,105]. In this context, metformin has been widely discussed as a candidate geroprotective agent, largely through AMPK activation and downstream inhibition of mTOR signaling, with additional links to autophagy and metabolic regulation[100,103]. Likewise, mTOR inhibition has been studied as a strategy to reproduce part of the biological effect of nutrient limitation[103,106]. At this point, these approaches are viewed as ways to explore mechanisms and potentially prevent adverse outcomes.
Extracellular matrix mechanics modulation
Gradual alterations in extracellular matrix composition and mechanical properties are a consistent feature of cardiac aging in the absence of overt disease[11]. Increased myocardial stiffness has been linked more closely to changes in collagen crosslinking and matrix organization than to large increases in collagen content[1]. Mechanical testing of septal biopsies showed that the increase in tangent modulus correlates with advanced glycation end-product (AGE)-mediated collagen cross-linking. This change is driven more by aberrant collagen cross-linking and altered matrix organization than by any substantial rise in total collagen content. In 24-month-old C57BL/6 mice, blunting collagen cross-linking enzymes or limiting the accumulation of advanced glycation end-products can partially restore tissue compliance and improve diastolic performance in aged rodents without reversing established structural remodeling[98,107]. During the aging process of a healthy heart, selectively modulating collagen quality (rather than reversing existing fibrosis) can preserve a compliant matrix and delay the onset of diastolic dysfunction.
Sustaining mitochondrial and metabolic adaptability
Even in clinically silent hearts, mitochondrial number, cristae density and respiratory chain efficiency fall by 20%-30 % between early adult life and 24 months of age in rodents, while reserve capacity declines twice as fast[107,108]. These structural changes are accompanied by a shift from fatty-acid β-oxidation toward carbohydrate oxidation, an adaptation that lowers oxygen cost per ATP but reduces the heart’s ability to switch substrates when workload suddenly rises. Because the production of basal ATP remains adequate, the energetic defect is only revealed under stress conditions. During rapid pacing or ischemia-reperfusion, aged mitochondria produce more reactive oxygen species (ROS) and less phosphocreatine, which leads to contractile decline[109]. In older adults, six weeks of nicotinamide riboside supplementation doubled whole-blood NAD+ and enhanced mitochondrial respiration in peripheral mononuclear cells, yet the accompanying reduction in systolic pressure was not accompanied by restoration of a youthful acyl-carnitine profile[110]. These observations indicate that supportive, rather than restorative, interventions can preserve metabolic adaptability and energetic reserve, thereby buffering the aging heart against cumulative stress.
In recent years, increasing attention has been directed toward interventions that preserve mitochondrial quality control[111]. This is particularly relevant in the aging heart, where impaired mitophagy and defective mitochondrial turnover contribute to the accumulation of dysfunctional mitochondria[112]. Eisenberg et al. showed that oral spermidine supplementation prolonged lifespan and conferred cardioprotective effects, including reduced cardiac hypertrophy and preserved diastolic function[113]. These benefits were associated with enhanced autophagy, mitophagy, and mitochondrial respiration. Therefore, in a review on cardiac aging, spermidine is best discussed as a biologically plausible intervention that may support intracellular quality-control pathways[114,115]. Urolithin A has attracted increasing interest as a mitophagy-promoting compound with potential relevance to cardiac aging[116]. Andreux et al. reported that urolithin A was safe in humans and induced a molecular profile consistent with improved mitochondrial and cellular health[117]. In addition, Huang et al. reported that urolithin A alleviated obesity-induced metabolic cardiomyopathy in mice, improved diastolic dysfunction and cardiac remodeling, and that these benefits were associated with restoration of mitophagy[118]. Recent human studies have shown that urolithin A is well tolerated and can improve biomarkers related to mitochondrial function, although these trials were conducted mainly in skeletal muscle or immune aging rather than in the heart[119]. This mechanism remains relevant to cardiac aging because impaired mitophagy is increasingly recognized as a contributor to age-related mitochondrial dysfunction in cardiomyocytes. Therefore, urolithin A may be discussed as a promising mitophagy-oriented candidate in cardiac aging.
These findings suggest that mitochondrial interventions in healthy aging may be valuable not because they restore a youthful metabolic state, but because they help preserve mitochondrial quality control and metabolic flexibility under stress. This may be especially important in the aging heart, where resting systolic function is often maintained while reserve capacity gradually declines.
CONCLUSIONS
Cardiac remodeling during healthy aging develops gradually under the combined influence of lifelong mechanical load, metabolic demand, and accumulated molecular changes. Rather than following a uniform or purely degenerative course, aging hearts commonly retain resting systolic function while showing concentric geometric adaptation, subtle alterations in diastolic performance, and a progressive reduction in functional reserve. Together, these features separate physiological aging from disease-driven remodeling. Mechanistically, the maintenance or decline of cardiac adaptive capacity depends on the functional integrity of multiple interconnected layers. Intercellular signaling, extracellular matrix mechanics, mitochondrial quality control, DNA damage responses, and epigenetic programs collectively shape myocardial stiffness, energetic flexibility, and responsiveness to stress. When these systems operate efficiently, they preserve functional reserve and stress tolerance; however, their age-related dysregulation progressively erodes adaptive capacity, lowering the threshold for functional decompensation. For instance, impaired mitochondrial and metabolic adaptability reduces energetic reserve, while epigenetic drift and DNA repair deficiency compromise transcriptional and genomic stress responses.
From a clinical standpoint, current data favor strategies that preserve functional reserve and adaptive capacity rather than attempts to reverse established structural features of the aging heart. Interventions such as structured exercise, selective modulation of extracellular matrix properties, and support of mitochondrial and metabolic adaptability show that cardiac resilience can be enhanced even when baseline geometry remains unchanged. Maintaining this adaptive potential may therefore represent a practical path toward sustaining cardiovascular function. Further advances will rely on linking mechanistic insights to longitudinal human observations in order to clarify when adaptive remodeling transitions into functional limitation. Defining these boundaries may guide the development of combined, mechanism-based approaches that delay maladaptive remodeling, preserve stress tolerance, and extend cardiovascular healthspan during aging.
DECLARATIONS
Acknowledgement
The Graphical Abstract was created using figdraw.com (Copyright Code: TRATYdd5b5).
Authors’ contributions
Wrote the manuscript: Shen Z, Zhang Y
Edited the manuscript: Tang Q
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Regional Innovation and Development Joint Fund of the National Natural Science Foundation of China (No. U22A20269).
Conflicts of interest
Tang Q is Associate Editor of The Journal of Cardiovascular Aging. Tang Q was not involved in any steps of editorial processing, notably including reviewers’ selection, manuscript handling and decision making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
The Author(s) 2026.
REFERENCES
1. Li X, Pang X, Sun H, et al. Cardiac aging: molecular mechanisms and therapeutic interventions. Pharmacol Res. 2025;221:107954.
2. Ribeiro ASF, Zerolo BE, López-Espuela F, Sánchez R, Fernandes VS. Cardiac system during the aging process. Aging Dis. 2023;14:1105-22.
4. Vetter VM, Drewelies J, Sommerer Y, et al. Epigenetic aging and perceived psychological stress in old age. Transl Psychiatry. 2022;12:410.
5. Gissler MC, Antiochos P, Ge Y, Heydari B, Gräni C, Kwong RY. Cardiac magnetic resonance evaluation of LV remodeling post-myocardial infarction: prognosis, monitoring and trial endpoints. JACC Cardiovasc Imaging. 2024;17:1366-80.
6. Leancă SA, Crișu D, Petriș AO, et al. Left ventricular remodeling after myocardial infarction: from physiopathology to treatment. Life. 2022;12:1111.
7. Subramanian V, Tucker WJ, Peters AE, Upadhya B, Kitzman DW, Pandey A. Cardiovascular aging and exercise: implications for heart failure prevention and management. Circ Res. 2025;137:205-30.
8. Cheng S, Fernandes VR, Bluemke DA, McClelland RL, Kronmal RA, Lima JA. Age-related left ventricular remodeling and associated risk for cardiovascular outcomes: the multi-ethnic study of atherosclerosis. Circ Cardiovasc Imaging. 2009;2:191-8.
9. Hernández-Vicente A, Hernando D, Santos-Lozano A, et al. Heart rate variability and exceptional longevity. Front Physiol. 2020;11:566399.
10. Zhao D, Wang Y, Wong ND, Wang J. Impact of aging on cardiovascular diseases: from chronological observation to biological insights: JACC family series. JACC Asia. 2024;4:345-58.
11. Tracy E, Rowe G, LeBlanc AJ. Cardiac tissue remodeling in healthy aging: the road to pathology. Am J Physiol Cell Physiol. 2020;319:C166-82.
13. Li M, Fukagawa NK. Age-related changes in redox signaling and VSMC function. Antioxid Redox Signal. 2010;12:641-55.
14. Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490-7.
15. Janosevic D, Bertman K, Roth M, et al. Left ventricular concentric remodeling in normal aging is associated with decline of diastolic function assessed by multi-modality imaging. J Cardiovasc Magn Reson. 2011;13:1932.
16. Pillai R, Zhang L, Peters K, et al. Age- and sex-differences and reference values for ventricular strain by cardiovascular magnetic resonance imaging in adults without cardiovascular disease or cardiovascular disease risk factors. J Cardiovasc Magn Reson. 2025;27:101902.
17. Thomas JD, Edvardsen T, Abraham T, et al. Clinical applications of strain echocardiography: a clinical consensus statement from the American Society of Echocardiography Developed in collaboration with the European Association of Cardiovascular Imaging of the European Society of Cardiology. Eur Heart J Cardiovasc Imaging. 2026;27:335-68.
18. Kersten J, Hackenbroch C, Bouly M, Tyl B, Bernhardt P. What is normal for an aging heart? A Prospective CMR Cohort Study. J Cardiovasc Imaging. 2022;30:202-11.[DOI:10.4250/jcvi.2022. 0021].
19. Zhai P, Sung EA, Shiheido-Watanabe Y, Takayama K, Tian Y, Sadoshima J. Suppression of autophagy induces senescence in the heart. bioRxiv. 2024. 2024. Available from: https://www.biorxiv.org/content/10.1101/2024.05.26.595978v1.abstract [Last accessed on 28 May 2026].
20. Hu WS, Liao WY, Chang CH, Chen TS. Paracrine IGF-1 activates SOD2 expression and regulates ROS/p53 axis in the treatment of cardiac damage in D-galactose-induced aging rats after receiving mesenchymal stem cells. J Clin Med. 2022;11:4419.
21. Conte M, Petraglia L, Poggio P, et al. Inflammation and cardiovascular diseases in the elderly: the role of epicardial adipose tissue. Front Med. 2022;9:844266.
22. Loffredo FS, Steinhauser ML, Jay SM, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828-39.
23. Egerman MA, Cadena SM, Gilbert JA, et al. GDF11 Increases with age and inhibits skeletal muscle regeneration. Cell Metab. 2015;22:164-74.
24. Kraler S, Balbi C, Vdovenko D, et al. Circulating GDF11 exacerbates myocardial injury in mice and associates with increased infarct size in humans. Cardiovasc Res. 2023;119:2729-42.
25. Ketchem JM, Bowman EJ, Isales CM. Male sex hormones, aging, and inflammation. Biogerontology. 2023;24:1-25.
26. Szabó R, Hoffmann A, Börzsei D, et al. Hormone replacement therapy and aging: a potential therapeutic approach for age-related oxidative stress and cardiac remodeling. Oxid Med Cell Longev. 2021;2021:8364297.
27. Morin-Grandmont A, Walsh-Wilkinson É, Labbé EA, et al. Biological sex, sex steroids and sex chromosomes contribute to mouse cardiac aging. Aging. 2024;16:7553-77.
28. Prud’homme GJ, Kurt M, Wang Q. Pathobiology of the klotho antiaging protein and therapeutic considerations. Front Aging. 2022;3:931331.
29. Chen K, Wang S, Sun QW, Zhang B, Ullah M, Sun Z. Correction to: klotho deficiency causes heart aging via impairing the Nrf2-GR pathway. Circ Res. 2025;137:e175.
30. Guo XH, Li YH, Zhao YS, Zhai YZ, Zhang LC. Antiaging effects of melatonin on the myocardial mitochondria of rats and associated mechanisms. Mol Med Rep. 2017;15:403-10.
32. Vidal R, Wagner JUG, Braeuning C, et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight. 2019;4:131092.
33. Chang ACY, Pardon G, Chang ACH, et al. Increased tissue stiffness triggers contractile dysfunction and telomere shortening in dystrophic cardiomyocytes. Stem Cell Rep. 2021;16:2169-81.
34. Schneider SE, Scott AK, Gallagher KM, Miller EY, Ghosh S, Neu CP. Mechanical stress triggers premature senescence in cardiac fibroblasts. Adv Sci. 2025;12:e13314.
35. Angelini A, Ortiz-Urbina J, Trial J, et al. Sex-specific phenotypes in the aging mouse heart and consequences for chronic fibrosis. Am J Physiol Heart Circ Physiol. 2022;323:H285-300.
36. Ji H, Kwan AC, Chen MT, et al. Sex differences in myocardial and vascular aging. Circ Res. 2022;130:566-77.
37. Zheng X, Yao G, Yu H, et al. FBLN7 KO attenuates age-related cardiac fibrosis by promoting TGFBR3/ALK1/Smad1 signaling and inhibiting the profibrotic phenotypes of cardiac fibroblasts. Theranostics. 2025;15:8531-52.
38. Wagner JUG, Dimmeler S. Cellular cross-talks in the diseased and aging heart. J Mol Cell Cardiol. 2020;138:136-46.
39. Vagnozzi RJ, Molkentin JD. Resident macrophages keep mitochondria running in the heart. Cell Res. 2020;30:1057-8.
40. Nicolás-Ávila JA, Lechuga-Vieco AV, Esteban-Martínez L, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. 2020;183:94-109.e23.
41. Adao DMT, Ching C, Fish JE, Simmons CA, Billia F. Endothelial cell-cardiomyocyte cross-talk: understanding bidirectional paracrine signaling in cardiovascular homeostasis and disease. Clin Sci. 2024;138:1395-419.
42. Gao C, Xia Y, Li C, et al. KMV-mediated cardiomyocyte-to-endothelial cell signaling drives capillary rarefaction to promote heart failure following pressure overload. Theranostics. 2025;15:4970-88.
43. Tamiato A, Tombor LS, Fischer A, et al. Age-dependent RGS5 loss in pericytes induces cardiac dysfunction and fibrosis. Circ Res. 2024;134:1240-55.
44. Simon C, Greening DW, Bolumar D, Balaguer N, Salamonsen LA, Vilella F. Extracellular vesicles in human reproduction in health and disease. Endocr Rev. 2018;39:292-332.
45. Grigorian Shamagian L, Rogers RG, Luther K, et al. Rejuvenating effects of young extracellular vesicles in aged rats and in cellular models of human senescence. Sci Rep. 2023;13:12240.
46. Chen X, Luo Y, Zhu Q, et al. Small extracellular vesicles from young plasma reverse age-related functional declines by improving mitochondrial energy metabolism. Nat Aging. 2024;4:814-38.
47. Aijaz A, Bieerkehazhi S, Kang N, et al. Young extracellular vesicles restore burn-induced adipose tissue immunometabolic and mitochondrial function in older mice. Sci Rep. 2025;15:35328.
48. Sanz-Ros J, Huete-Acevedo J, Mas-Bargues C, et al. Small extracellular vesicles from young adipose-derived stem cells ameliorate age-related changes in the heart of old mice. Stem Cell Res Ther. 2025;16:138.
49. Saeedi Saravi SS, Bonetti NR, Vukolic A, et al. Long-term dietary n3 fatty acid prevents aging-related cardiac diastolic and vascular dysfunction. Vascul Pharmacol. 2023;150:107175.
50. Abdellatif M, Sedej S, Kroemer G. NAD+ metabolism in cardiac health, aging, and disease. Circulation. 2021;144:1795-817.
51. Santulli G, Kansakar U, Varzideh F, Mone P, Jankauskas SS, Lombardi A. Functional role of taurine in aging and cardiovascular health: an updated overview. Nutrients. 2023;15:4236.
52. Tang X, Li PH, Chen HZ. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front Endocrinol 2020;11:280.
53. Morgado LAL, Rodrigues LMZ, Silva DCF, da Silva BD, Irigoyen MCC, Takano APC. NF-κB-specific suppression in cardiomyocytes unveils aging-associated responses in cardiac tissue. Biomedicines. 2025;13:224.
54. Yousef A, Fang L, Heidari M, Kranrod J, Seubert JM. The role of CYP-sEH derived lipid mediators in regulating mitochondrial biology and cellular senescence: implications for the aging heart. Front Pharmacol. 2024;15:1486717.
55. De Bartolo A, Rago V, Romeo N, et al. Unveiling selenoprotein T as a novel regulator of cardiomyocyte senescence: pivotal role of the CD36 receptor in AC16 human cardiomyocytes. GeroScience. 2026;48:1751-70.
56. de Boer M, Te Lintel Hekkert M, Chang J, et al. DNA repair in cardiomyocytes is critical for maintaining cardiac function in mice. Aging Cell. 2023;22:e13768.
57. Jiang R, Wang H, Zhang W, et al. DHT ameliorates cardiac aging in progeroid mice by XRCC4-mediated genome stabilization. Mech Ageing Dev. 2026;229:112141.
58. Cai Y, Liu H, Song E, et al. Deficiency of telomere-associated repressor activator protein 1 precipitates cardiac aging in mice via p53/PPARα signaling. Theranostics. 2021;11:4710-27.
59. Maggiorani D, Santin Y, Formoso K, et al. Identification of Prominin-2 as a new player of cardiomyocyte senescence in the aging heart. Aging Cell. 2024;23:e14204.
60. Anderson R, Lagnado A, Maggiorani D, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019;38.
61. Qian W, Kumar N, Roginskaya V, et al. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc Natl Acad Sci U S A. 2019;116:18435-44.
62. Li B, Xiong W, Zuo W, et al. Proximal telomeric decompaction due to telomere shortening drives FOXC1-dependent myocardial senescence. Nucleic Acids Res. 2024;52:6269-84.
63. Calvani R, Joseph AM, Adhihetty PJ, et al. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol Chem. 2013;394:393-414.
64. Pan G, Deshpande M, Pang H, et al. 4-Hydroxy-2-nonenal attenuates 8-oxoguanine DNA glycosylase 1 activity. J Cell Biochem. 2020;121:4887-97.
65. Rong Z, Tu P, Xu P, et al. The mitochondrial response to DNA damage. Front Cell Dev Biol. 2021;9:669379.
66. Manolis AS, Manolis AA, Manolis TA, et al. Mitochondrial dysfunction in cardiovascular disease: current status of translational research/clinical and therapeutic implications. Med Res Rev. 2021;41:275-313.
67. Elorza AA, Soffia JP. mtDNA heteroplasmy at the core of aging-associated heart failure. An integrative view of OXPHOS and mitochondrial life cycle in cardiac mitochondrial physiology. Front Cell Dev Biol. 2021;9:625020.
68. Sato M, Kadomatsu T, Morinaga J, et al. HINT1 suppression protects against age-related cardiac dysfunction by enhancing mitochondrial biogenesis. Mol Metab. 2025;93:102107.
69. Guo Y, You Y, Shang FF, et al. iNOS aggravates pressure overload-induced cardiac dysfunction via activation of the cytosolic-mtDNA-mediated cGAS-STING pathway. Theranostics. 2023;13:4229-46.
70. Gabillard-Lefort C, Thibault T, Lenaers G, Wiesner RJ, Mialet-Perez J, Baris OR. Heart of the matter: mitochondrial dynamics and genome alterations in cardiac aging. Mech Ageing Dev. 2025;224:112044.
71. Yan M, Li Y, Luo Q, et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. 2022;8:258.[DOI:10. 1038/s41420-022-01046-w].
72. Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145-53.
73. Glancy B, Hartnell LM, Malide D, et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature. 2015;523:617-20.
74. Glancy B, Hartnell LM, Combs CA, et al. Power grid protection of the muscle mitochondrial reticulum. Cell Rep. 2017;19:487-96.
75. El'darov ChM, Vays VB, Vangeli IM, Kolosova NG, Bakeeva LE. Morphometric examination of mitochondrial ultrastructure in aging cardiomyocytes. Biochemistry 2015;80:604-9.
76. Picca A, Lezza AM. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion. 2015;25:67-75.
77. Zhang Y, Wang C, Zhou J, et al. Complex inhibition of autophagy by mitochondrial aldehyde dehydrogenase shortens lifespan and exacerbates cardiac aging. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1919-32.
78. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res. 2016;118:1563-76.
79. Zhan S, Guo C, Yan H, Zheng G, Yan D. The multi-dimensional regulatory mechanism of Sirt6 in heart health: From cell death pathways to targeted therapy for cardiovascular diseases. Biochem Biophys Res Commun. 2025;782:152561.
80. Delbridge LMD, Mellor KM, Taylor DJ, Gottlieb RA. Myocardial stress and autophagy: mechanisms and potential therapies. Nat Rev Cardiol. 2017;14:412-25.
81. Hsu YJ, Hsu SC, Hsu CP, et al. Sirtuin 1 protects the aging heart from contractile dysfunction mediated through the inhibition of endoplasmic reticulum stress-mediated apoptosis in cardiac-specific Sirtuin 1 knockout mouse model. Int J Cardiol. 2017;228:543-52.
82. Kerrigan L, Edgar K, Russell-Hallinan A, et al. Integrin beta-like 1 is regulated by DNA methylation and increased in heart failure patients. ESC Heart Fail. 2025;12:150-65.
83. Si J, Chen L, Yu C, et al. ; China Kadoorie Biobank Collaborative Group. Healthy lifestyle, DNA methylation age acceleration, and incident risk of coronary heart disease. Clin Epigenetics. 2023;15:52.
84. Topriceanu CC, Dev E, Ahmad M, et al. Accelerated DNA methylation age plays a role in the impact of cardiovascular risk factors on the human heart. Clin Epigenetics. 2023;15:164.
85. Serio S, Pagiatakis C, Musolino E, et al. Cardiac aging is promoted by pseudohypoxia increasing p300-induced glycolysis. Circ Res. 2023;133:687-703.
86. Grzeczka A, Graczyk S, Gong X, Gröschel J, Spethmann S, Kordowitzki P. Aging hearts, fibrotic fears: The sirtuin connection. Biomed Pharmacother. 2025;193:118882.
87. Trembinski DJ, Bink DI, Theodorou K, et al. Aging-regulated anti-apoptotic long non-coding RNA Sarrah augments recovery from acute myocardial infarction. Nat Commun. 2020;11:2039.
88. Cabiati M, Sapio A, Salvadori C, et al. Evaluation of transcriptional levels of the natriuretic peptides, endothelin-1, adrenomedullin, their receptors and long non-coding RNAs in rat cardiac tissue as cardiovascular biomarkers of aging. Peptides. 2020;123:170173.
89. Jha S, Thasma Loganathbabu VK, Kumaran K, Krishnasamy G, Aruljothi KN. Long non-coding RNAs (lncRNAs) in heart failure: a comprehensive review. Noncoding RNA. 2023;10:3.
90. Lin R, Rahtu-Korpela L, Magga J, et al. miR-1468-3p promotes aging-related cardiac fibrosis. Mol Ther Nucleic Acids. 2020;20:589-605.
91. Pandey A, Kraus WE, Brubaker PH, Kitzman DW. Healthy aging and cardiovascular function: invasive hemodynamics during rest and exercise in 104 healthy volunteers. JACC Heart Fail. 2020;8:111-21.
92. Jefferis BJ, Parsons TJ, Sartini C, et al. Objectively measured physical activity, sedentary behaviour and all-cause mortality in older men: does volume of activity matter more than pattern of accumulation? Br J Sports Med 2019;53:1013-20.
93. Lin R, Rahtu-Korpela L, Magga J, et al. miR-1468-3p promotes aging-related cardiac fibrosis. Mol Ther Nucleic Acids. 2020;20:589-605.
94. Borlaug BA, Nishimura RA, Sorajja P, Lam CS, Redfield MM. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail. 2010;3:588-95.
95. Howden EJ, Sarma S, Lawley JS, et al. Reversing the cardiac effects of sedentary aging in middle age-a randomized controlled trial: implications for heart failure prevention. Circulation. 2018;137:1549-60.
96. Beaumont AJ, Grace FM, Richards JC, Campbell AK, Sculthorpe NF. Aerobic training protects cardiac function during advancing age: a meta-analysis of four decades of controlled studies. Sports Med. 2019;49:199-219.
97. Fleg JL, Morrell CH, Bos AG, et al. Accelerated longitudinal decline of aerobic capacity in healthy older adults. Circulation. 2005;112:674-82.
98. Roh JD, Houstis N, Yu A, et al. Exercise training reverses cardiac aging phenotypes associated with heart failure with preserved ejection fraction in male mice. Aging Cell. 2020;19:e13159.
99. Green CL, Lamming DW, Fontana L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat Rev Mol Cell Biol. 2022;23:56-73.
100. Radovic M, Gartzke LP, Wink SE, van der Kleij JA, Politiek FA, Krenning G. Targeting the electron transport system for enhanced longevity. Biomolecules. 2025;15:614.
101. Niemann B, Li L, Simm A, et al. Caloric restriction reduces sympathetic activity similar to beta-blockers but conveys additional mitochondrio-protective effects in aged myocardium. Sci Rep. 2021;11:1931.
102. Koutouroushis C, Sarkar O. Role of autophagy in cardiovascular disease and aging. Cureus. 2021;13:e20042.
103. Murillo-Cancho AF, Lozano-Paniagua D, Nievas-Soriano BJ. Dietary and pharmacological modulation of aging-related metabolic pathways: molecular insights, clinical evidence, and a translational model. Int J Mol Sci. 2025;26:9643.
104. Small S, Iglesies-Grau J, Gariepy C, Wilkinson M, Taub P, Kirkham A. Time-restricted eating: a novel dietary strategy for cardiac rehabilitation. Can J Cardiol. 2023;39:S384-94.
105. Gabel K, Cienfuegos S, Kalam F, Ezpeleta M, Varady KA. Time-restricted eating to improve cardiovascular health. Curr Atheroscler Rep. 2021;23:22.
106. Chaudhary S, Chaudhary MR, Jena MK, et al. Calorie restriction mimetics against aging and inflammation. Biogerontology. 2025;26:126.
107. Zhang Y, Mi SL, Hu N, et al. Mitochondrial aldehyde dehydrogenase 2 accentuates aging-induced cardiac remodeling and contractile dysfunction: role of AMPK, Sirt1, and mitochondrial function. Free Radic Biol Med. 2014;71:208-20.
108. Makino N, Maeda T. Calorie restriction delays cardiac senescence and improves cardiac function in obese diabetic rats. Mol Cell Biochem. 2021;476:221-9.
109. Weisel FJ, Mullett SJ, Elsner RA, et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat Immunol. 2020;21:331-42.
110. Wang DD, Airhart SE, Zhou B, et al. Safety and tolerability of nicotinamide riboside in heart failure with reduced ejection fraction. JACC Basic Transl Sci. 2022;7:1183-96.
111. Ali MA, Gioscia-Ryan R, Yang D, Sutton NR, Tyrrell DJ. Cardiovascular aging: spotlight on mitochondria. Am J Physiol Heart Circ Physiol. 2024;326:H317-33.
112. Wang S, Long H, Hou L, et al. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther. 2023;8:304.
113. Eisenberg T, Abdellatif M, Schroeder S, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428-38.
114. Hofer SJ, Simon AK, Bergmann M, Eisenberg T, Kroemer G, Madeo F. Mechanisms of spermidine-induced autophagy and geroprotection. Nat Aging. 2022;2:1112-29.
115. Hofer SJ, Daskalaki I, Bergmann M, et al. Spermidine is essential for fasting-mediated autophagy and longevity. Nat Cell Biol. 2024;26:1571-84.
116. D’Amico D, Andreux PA, Valdés P, Singh A, Rinsch C, Auwerx J. Impact of the natural compound urolithin a on health, disease, and aging. Trends Mol Med. 2021;27:687-99.
117. Andreux PA, Blanco-Bose W, Ryu D, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1:595-603.
118. Huang JR, Zhang MH, Chen YJ, et al. Urolithin A ameliorates obesity-induced metabolic cardiomyopathy in mice via mitophagy activation. Acta Pharmacol Sin. 2023;44:321-31.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.