

Electrochemical alkene isomerization enables carboxylation of unactivated alkenes with CO2
 0
0 
Keywords
Carbon dioxide (CO2) is widely employed as a versatile C1 feedstock in chemical synthesis owing to its non-toxic nature, affordability, and renewable availability[1]. Recently, various robust methodologies for converting CO2 into carboxylic acids[2-5] have been developed. However, the efficient conversion of CO2 under mild conditions remains challenging due to its high activation barrier, which typically requires substantial energy input. To this end, diverse methods spanning thermochemical, photochemical, biological, and electrochemical techniques have been established to transform CO2 into value-added products[6-8]. Among these, electrochemistry offers a transformative and scalable pathway for CO2 valorization, distinguished by its mild operating conditions and compatibility with renewable electricity[9,10]. A prominent example is the electrochemical carboxylation of alkenes with CO2, which enables the efficient synthesis of functionalized carboxylic acids from simple feedstocks[11]. To date, most established electrochemical protocols have primarily focused on activated alkenes, such as styrenes and conjugated dienes, which exhibit higher reactivity toward CO2 incorporation[5,12-15]. In contrast, unactivated alkenes remain challenging substrates for electrochemical carboxylation, mainly due to their high reduction potentials and the instability of the corresponding radical intermediates[16,17].
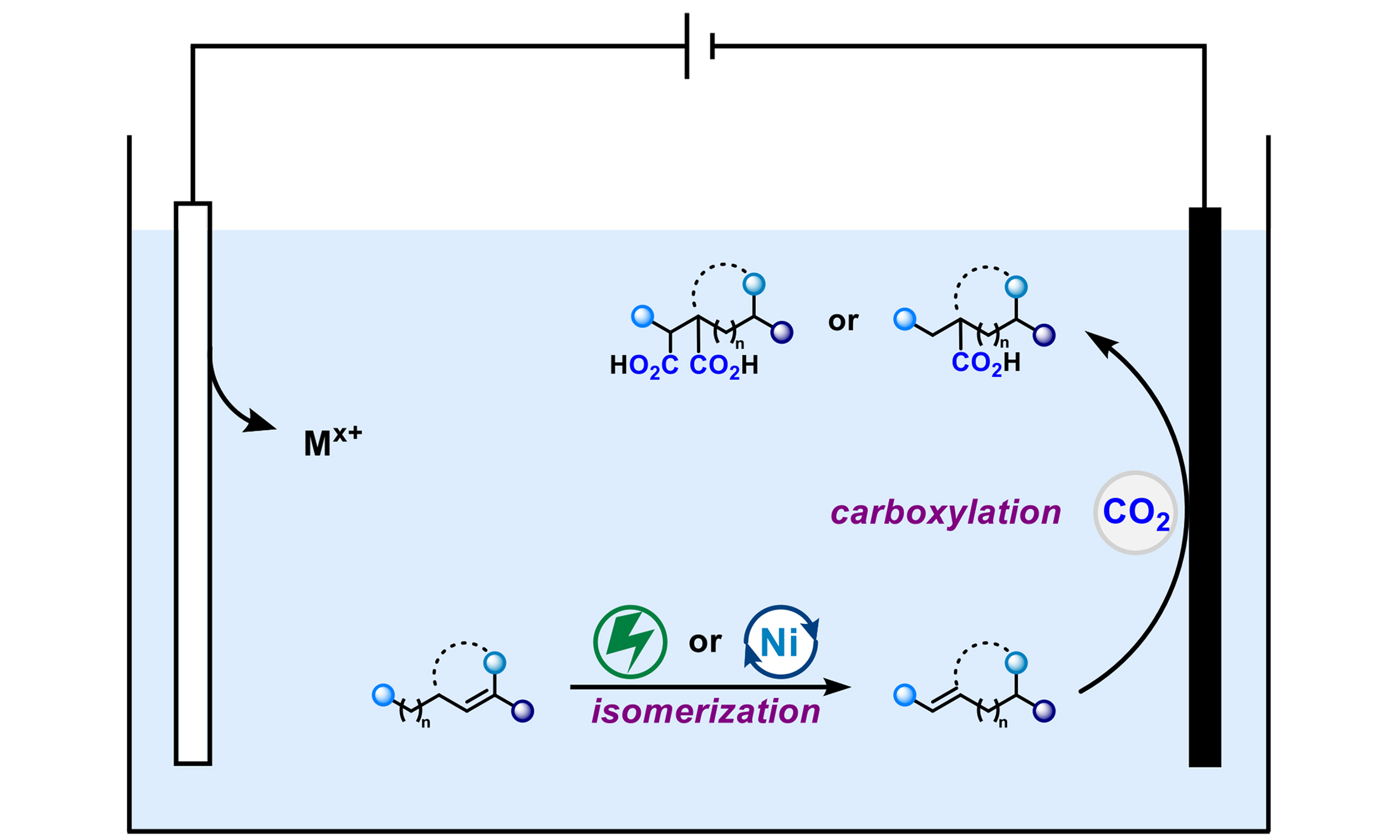
To tackle this issue, Dong et al. have recently developed two distinct methodologies based on a shared core strategy - (nickel-)electrochemical alkene isomerization[18,19] - to achieve the carboxylation of unactivated alkenes [Figure 1A]. Specifically, the process begins with a key electrochemical alkene isomerization step that converts non-conjugated alkenes into their conjugated counterparts under mild conditions. The resulting conjugated intermediates then undergo efficient tandem electroreductive carboxylation with CO2. This strategy relies on a critical pre-activation stage that transforms unreactive non-conjugated alkenes into thermodynamically more favorable conjugated isomers for subsequent carboxylation. This “isomerization-first” paradigm effectively overcomes the intrinsic limitations of unactivated alkenes, offering a general and powerful solution that significantly expands the scope of electrochemical CO2 utilization beyond activated substrates.
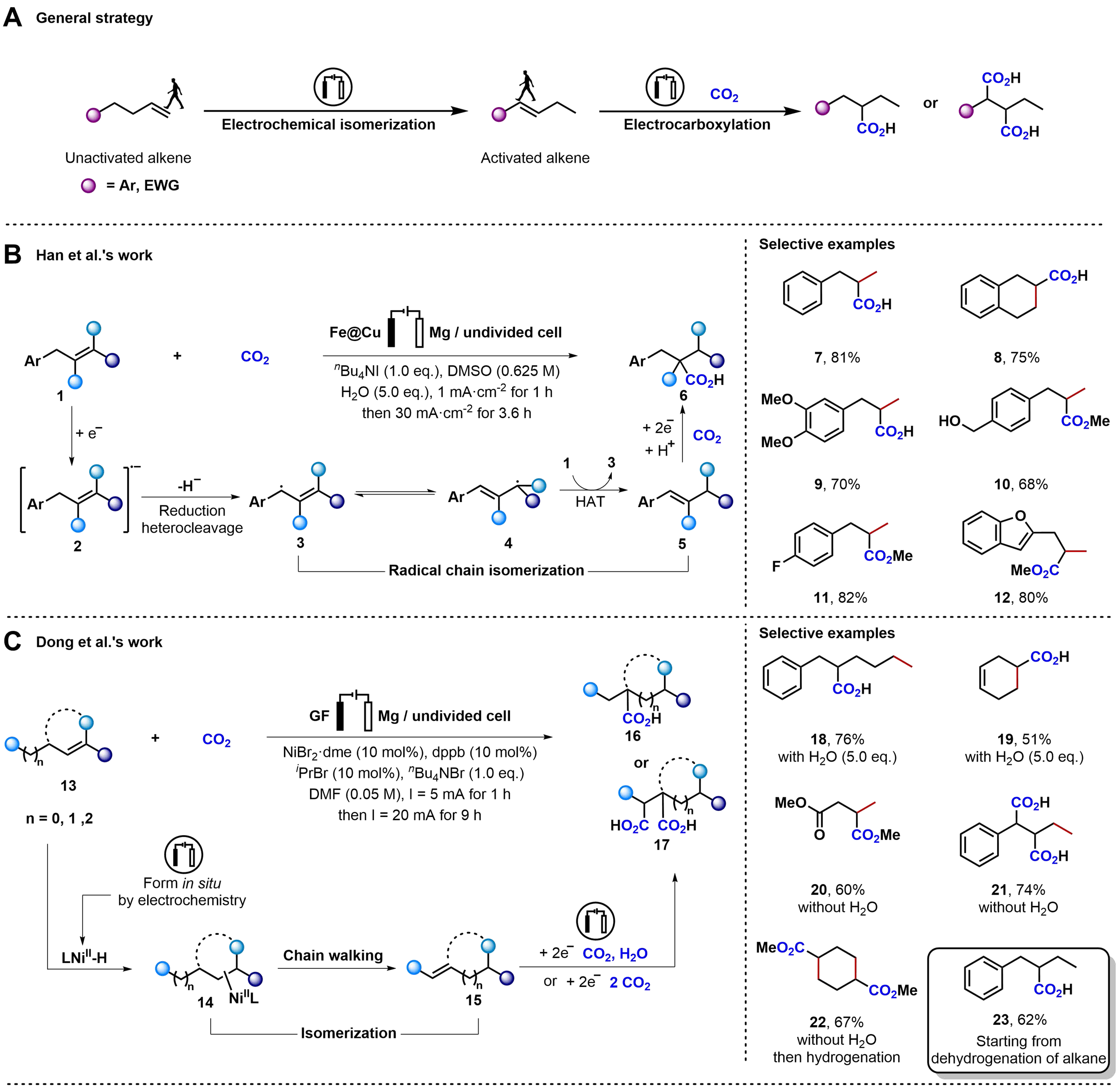
Figure 1. Electrochemical strategies for the carboxylation of unactivated alkenes with CO2 via pre-activation through alkene isomerization. (A) General strategy; (B) Electrocarboxylation via cathode-induced alkene isomerization; (C) Electrocarboxylation via metal-mediated alkene isomerization. EWG: Electron withdrawing group; Fe@Cu: Fe-electroplated Cu cathode; DMSO: dimethyl sulfoxide; HAT: hydrogen atom transfer; GF: graphite felt; dppb: 1,4-bis(diphenylphosphino)butane; DMF: N,N-dimethylformamide.
In Dong et al.’s work [Figure 1B][18], an electroreductive strategy for alkene isomerization was developed by directly activating the C–H bond of alkene 1, generating intermediate 2 along with a carbon radical 3 and a hydride ion. The carbon radical 3 then tautomerizes to carbon radical 4, followed by a hydrogen atom transfer process with 1 to yield the conjugated alkene 5. This key step enables the metal-free isomerization of unactivated alkenes into conjugated alkenes via C–H activation under mild conditions. Finally, the resulting conjugated alkene 5 undergoes electroreductive hydrocarboxylation with CO2 and water in a tandem process. This protocol exhibits a broad substrate scope, and a range of allylbenzene derivatives including both acyclic and cyclic substrates (7-12) undergo alkene isomerization and hydrocarboxylation smoothly. Moreover, some functional groups or heterocyclic moieties that are typically sensitive to redox environments, such as unprotected hydroxy groups (10), fluoro substituents (11), and benzofuran (12), are well tolerated under these conditions, demonstrating the broad applicability of this method.
Very recently, Dong et al. developed a nickel (Ni)-catalyzed system to achieve selective isomerization–carboxylation of unactivated alkenes [Figure 1C][19]. In this protocol, the Ni–H species is formed in situ via electrochemistry and serves as a key mediator for the isomerization of unactivated alkene 13 through a chain-walking mechanism, efficiently generating the activated conjugated alkenes 15. Subsequently, conjugated alkene 15 undergoes electroreductive carboxylation under a CO2 atmosphere to afford either monocarboxylic acid 16 or dicarboxylic acid 17 in a controllable manner, demonstrating the feasibility of both ectopic and remote C–H bond carboxylation. This protocol features a broad substrate scope and good functional group tolerance. A variety of functionalized acyclic and cyclic unactivated alkenes are well compatible and can be transformed into the corresponding carboxylic acids in satisfactory yields (18-22). Notably, alkane substrates can also serve as viable candidates for C–H carboxylation when alkane dehydrogenation is coupled with this tandem carboxylation strategy in a one-pot process, affording carboxylic acid 23 in 62% yield and further demonstrating the applicability of this protocol.
These two strategies offer distinct approaches, each with inherent trade-offs. The metal-free system benefits from operational simplicity and avoids the use of metal catalysts, which may be preferable for cost-sensitive or metal-free applications. In contrast, the Ni-catalyzed system generally provides higher efficiency and greater controllability, particularly for remote functionalization via chain walking, albeit at the cost of introducing a metal catalyst[20]. The choice between these strategies, therefore, depends on priorities such as selectivity, step economy, and metal compatibility.
In summary, the two complementary studies by Dong et al. elegantly showcase robust electrochemical strategies for the carboxylation of unactivated alkenes with CO2. Both methods operate under mild conditions and exhibit remarkable functional group tolerance and broad substrate compatibility, effectively addressing the long-standing challenge of carboxylating unactivated alkenes with CO2. Nevertheless, several challenges remain to be addressed in future studies. For instance, achieving precise control over regioselectivity, especially in highly functionalized or stereochemically complex alkenes, still requires further catalyst and reaction design. Future efforts could focus on extending this methodology to other challenging substrate classes, such as internal alkenes, long-chain aliphatic alkenes, or even alkynes, thereby further broadening the scope of electrocarboxylation. Additionally, the development of asymmetric variants, enabled by chiral catalysts or electrolytes, could provide access to enantiomerically enriched carboxylic acids, which are highly valuable in pharmaceutical and agrochemical synthesis. The integration of this electrochemical approach with continuous flow systems or renewable energy sources also holds promise for scaling up the process toward industrial applications, thereby contributing to a greener and more efficient carbon economy. Collectively, these advances substantially expand the synthetic toolbox for accessing valuable carboxylic acids from abundant feedstocks, providing a robust and practical platform for sustainable chemical synthesis.
DECLARATIONS
Authors’ contributions
Wrote the draft manuscript: Wang, B. N.; Sun, G. Q.
Revised and edited the manuscript: Yu, D. G.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This study was financially supported by the National Natural Science Foundation of China (22225106) and the Fundamental Research Funds for the Central Universities.
Conflicts of interest
Yu, D. G. serves as a Guest Editor of the Special Issue “Catalysis for CO2 Utilization” and as an Associate Editor of the journal Chemical Synthesis. Yu, D. G. was not involved in any steps of the editorial process, notably including reviewer selection, manuscript handling, or decision making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933.
2. Tortajada, A.; Juliá-Hernández, F.; Börjesson, M.; Moragas, T.; Martin, R. Transition-metal-catalyzed carboxylation reactions with carbon dioxide. Angew. Chem. Int. Ed. Engl. 2018, 57, 15948-82.
3. Yu, Z.; Shi, M. Recent advances in the electrochemically mediated chemical transformation of carbon dioxide. Chem. Commun. 2022, 58, 13539-55.
4. Jia, S.; Dong, M.; Zhu, Q.; Kang, X.; Wu, H.; Han, B. Electrochemical conversion of CO2 via C–X bond formation: recent progress and perspective. Chem. Synth. 2024, 4, 60.
5. Sun, G. Q.; Liao, L. L.; Ran, C. K.; Ye, J. H.; Yu, D. G. Recent advances in electrochemical carboxylation with CO2. Acc. Chem. Res. 2024, 57, 2728-45.
6. Qiu, L. Q.; Li, H. R.; He, L. N. Incorporating catalytic units into nanomaterials: rational design of multipurpose catalysts for CO2 valorization. Acc. Chem. Res. 2023, 56, 2225-40.
7. Chang, X.; Wang, T.; Yang, P.; Zhang, G.; Gong, J. The development of cocatalysts for photoelectrochemical CO2 reduction. Adv. Mater. 2019, 31, e1804710.
8. O’Brien, C. P.; Miao, R. K.; Shayesteh, Zeraati. A.; Lee, G.; Sargent, E. H.; Sinton, D. CO2 electrolyzers. Chem. Rev. 2024, 124, 3648-93.
9. Senboku, H. Electrochemical fixation of carbon dioxide: synthesis of carboxylic acids. Chem. Rec. 2021, 21, 2354-74.
10. Wang, Q.; Wang, Y.; Liu, M.; Chu, G.; Qiu, Y. Recent advances in photochemical/electrochemical carboxylation of olefins with CO2. Chin. J. Chem. 2024, 42, 2249-66.
11. Liu, X.; Zhang, K.; Tao, L.; Lu, X.; Zhang, W. Recent advances in electrochemical carboxylation reactions using carbon dioxide. Green. Chem. Eng. 2022, 3, 125-37.
12. Kim, Y.; Park, G. D.; Balamurugan, M.; Seo, J.; Min, B. K.; Nam, K. T. Electrochemical β-selective hydrocarboxylation of styrene using CO2 and water. Adv. Sci. 2020, 7, 1900137.
13. Alkayal, A.; Tabas, V.; Montanaro, S.; Wright, I. A.; Malkov, A. V.; Buckley, B. R. Harnessing applied potential: selective β-hydrocarboxylation of substituted olefins. J. Am. Chem. Soc. 2020, 142, 1780-5.
14. Xie, P.; Shi, A.; Qiu, Y. Electrochemical β-selective silylcarboxylation of styrenes with CO2. Sci. Bull. 2025, 70, 2411-5.
15. Bazzi, S.; Hu, L.; Schulz, E.; Mellah, M. Electrogenerated Sm(II)-catalyzed carbon dioxide reduction for β-hydrocarboxylation of styrenes. Organometallics 2023, 42, 1425-31.
16. Zhang, Y.; Zhao, X.; Qing, G. Electrochemical-induced hydrofunctionalizations of alkenes and alkynes. Chem. Synth. 2024, 4, 16.
17. Alektiar, S. N.; Han, J.; Dang, Y.; Rubel, C. Z.; Wickens, Z. K. Radical hydrocarboxylation of unactivated alkenes via photocatalytic formate activation. J. Am. Chem. Soc. 2023, 145, 10991-7.
18. Dong, M.; Jia, S.; Chen, X.; et al. Cathode-induced C-H bond heterolysis for olefin isomerization and applications in electrocarboxylation. J. Am. Chem. Soc. 2025, 147, 19976-85.
19. Dong, M.; Chen, H.; Liu, J.; et al. Electrocatalytic migratory carboxylation of unactivated olefins or halides with CO2. Angew. Chem. Int. Ed. Engl. 2026, 65, e18160.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.