


Hepatocellular carcinoma tumor immune microenvironment: heterogeneity and therapeutic strategies
 0
0 
Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related mortality worldwide, with immunotherapy emerging as a pivotal strategy for advanced disease. However, treatment responses remain highly variable, largely due to the complex and dynamic tumor immune microenvironment (TIME). This review systematically examines the composition, heterogeneity, and function of the TIME of HCC, focusing on the interplay between immunosuppressive cells - such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs) - and functional immune cells, including CTLs, NK cells, and DCs. We highlight the spatial and temporal heterogeneity of the TIME, shaped by underlying HCC etiologies, which critically influences immune evasion and therapeutic outcomes. We also evaluate current immunotherapeutic approaches, particularly immune checkpoint inhibitors (ICIs), adoptive cell therapies, and strategies targeting metabolic and microbial remodeling of the TIME. Finally, we discuss emerging combination therapies and future directions aimed at overcoming immunosuppressive barriers to enhance personalized treatment and improve clinical outcomes in HCC. A deeper understanding of TIME biology is essential for developing more effective immunotherapeutic strategies.
Keywords
INTRODUCTION
Hepatocellular carcinoma (HCC) remains a leading cause of global cancer mortality, largely due to its frequent late diagnosis and high recurrence rate following curative treatments[1,2]. In recent years, immunotherapy has emerged as a pivotal advancement in managing advanced HCC, with immune checkpoint inhibitors (ICIs) demonstrating notable clinical promise, particularly in combination regimens[3-6].
However, the efficacy of immunotherapy in HCC is highly variable, and only a subset of patients achieves a durable response. A major determinant of this heterogeneous treatment outcome is the complex and dynamic tumor immune microenvironment (TIME)[7]. The TIME of HCC is characterized by an intricate network of immune cells, stromal components, cytokines, and signaling molecules that collectively promote tumor progression, immune evasion, and therapeutic resistance[7]. Its significant spatial and temporal heterogeneity - influenced by etiology, disease stage, and host factors - plays a critical role in shaping immunosuppressive landscapes and limiting the success of immunotherapies[8,9].
To address these challenges, we herein propose an Integrated Three-Dimensional Heterogeneity Model that reframes the HCC TIME through the interplay of Etiology, Spatial Topology, and Temporal Evolution. Specifically, distinct pathogenic drivers dictate the foundational “immunological soil” and exhaustion trajectories; the physical stromal barriers and metabolic compartmentalization between the tumor core and invasive margin construct complex spatial networks of immune exclusion; and the TIME functions as a highly plastic system undergoing continuous adaptive remodeling during natural progression and clinical interventions.
By synthesizing these dimensions into a comprehensive conceptual framework, this review systematically elucidates how therapeutic strategies can be anchored to specific microenvironmental profiles - namely, through etiology-specific stratification, combination therapies designed to dismantle spatial barriers, and the strategic identification of “therapeutic windows” in immune remodeling. Ultimately, this multidimensional roadmap aims to provide a theoretical blueprint for reversing immune tolerance and achieving next-generation precision immunotherapy in HCC.
OVERALL COMPOSITION OF THE MICROENVIRONMENT OF HCC
From a literal standpoint, the tumor microenvironment (TME) broadly defined refers to the milieu surrounding a tumor. It comprises all non-malignant host cells in the vicinity, such as endothelial cells, diverse immune cells, cancer-associated fibroblasts (CAFs), and the signaling molecules - including growth factors, cytokines, chemokines, and extracellular vesicles - secreted by these cells. Furthermore, it encompasses the extracellular matrix (ECM) as well as the vascular and lymphatic networks[10]. Tumor formation, growth, invasion, and metastasis all rely on the TME. Conversely, the TME itself responds to extracellular signals released by the tumor, resulting in a dynamic, bidirectional interaction between the two[11].
The liver is a unique immune organ characterized by a distinct dual blood supply, which predisposes it to heightened susceptibility to viral/bacterial infections, tumors, and sterile tissue injury. Upon infection, the liver’s inherent immune tolerance is disrupted, triggering inflammatory responses. A rapid and potent innate immune response is first activated to clear pathogens. However, in cases of chronic infection such as hepatitis B Virus (HBV), the interplay between host adaptive immunity and viral immune-evasion mechanisms can lead to persistent, lifelong disease[12,13].
Hepatic immunity involves both resident and recruited immune cells. The liver harbors the body’s largest populations of natural killer (NK) cells and Kupffer cells (KCs), the latter being the largest group of tissue-resident macrophages. It is also enriched with natural killer T cells (NKT cells), which collectively form a robust innate immune compartment. Nevertheless, many immune responses in the liver depend on the recruitment of circulating immune cells, particularly neutrophils and monocytes[14-16].
Under physiological conditions, the liver maintains a state of immune tolerance, exhibiting low immune reactivity. This is partly due to continuous low-level exposure to gut-derived pathogen-associated molecular patterns (e.g., lipopolysaccharides), which dampen KC-mediated lymphocyte activation and help prevent excessive immune-mediated tissue damage. In pathological states - such as chronic viral hepatitis, alcoholic liver disease, and non-alcoholic fatty liver disease - this normal immune tolerance becomes fragile and easily breached[17-19].
Research indicates that persistent chronic stimulation, leading to irreversible processes like liver fibrosis or cirrhosis, can promote HCC development even after the primary stimulus (e.g., HCV) is eliminated. In such settings, HCC often arises in the context of pre-existing fibrosis or cirrhosis, suggesting that virus clearance may not fully reverse long-term alterations in the hepatic microenvironment, leaving the tissue prone to subsequent oncogenic events[17-19]. These changes persist through the formation of a precancerous microenvironment, which disrupts the balance between immune activation and suppression. A shift in the ratio of pro-inflammatory to anti-inflammatory cytokines is a hallmark of this stage, fostering a sustained pro-inflammatory milieu.
As the precancerous microenvironment evolves, malignant transformation occurs, and the liver’s immune-suppressive environment begins to protect nascent tumor cells. Immune evasion, tissue remodeling, and chronic inflammation persist, driving the transition from a precancerous to a tumor-supportive microenvironment[20]. Concurrently, immune cell recruitment leads to the infiltration of immunosuppressive populations - such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) - into TME. These cells further promote an immunosuppressive niche that facilitates cancer progression.
THE ROLE OF IMMUNOSUPPRESSIVE CELL POPULATIONS IN HCC
In HCC, beyond the intrinsic malignant properties of tumor cells, TIME actively enables immune evasion, allowing tumors to escape host immune surveillance and destruction. Key to this process are specific immunosuppressive cell populations within the TIME, most notably TAMs, MDSCs, and Tregs. These cells promote tumor progression, metastasis, and therapy resistance by secreting immunosuppressive factors, reshaping the immune landscape, and directly inhibiting effector immune cells such as T cells and NK cells.
TAMs/M2-like macrophages
TAMs predominantly exhibit an M2-like phenotype, primarily arising from the phenotypic transformation of resident KCs during tumor progression. Unlike pro-inflammatory, anti-tumor M1 macrophages, M2-polarized TAMs secrete anti-inflammatory factors such as Interleukin-10 (IL-10), transforming growth factor-β (TGF-β), and arginase-1 (Arg1), which suppress immune cell activity and foster an immunosuppressive microenvironment conducive to tumor growth[21]. Furthermore, M2 TAMs promote tumor progression by secreting angiogenic factors like vascular endothelial growth factor (VEGF) and chemokines such as CCL2, thereby stimulating vasculature formation and enhancing the metastatic potential of cancer cells[21,22].
In summary, TAMs are extensively infiltrated within HCC, and their polarization state is closely linked to tumor aggressiveness, immune evasion, and patient prognosis. Through the secretion of immunosuppressive factors, metabolic reprogramming, promotion of angiogenesis, and facilitation of metastasis, M2-like TAMs serve as a central driver of immune tolerance and malignant progression in HCC, making them a pivotal target for immunotherapy[23-26].
Source of TAMs and regulation of M2 polarization
TAMs in HCC originate from diverse sources, primarily including bone marrow-derived monocytes, splenic hematopoietic stem cell populations, and the local activation and phenotypic conversion of resident liver macrophages (KCs)[26-28]. These precursor cells are recruited to the tumor tissue by chemotactic signals within the TIME and undergo M2-like polarization under the influence of various regulatory factors.
This polarization is driven by both cytokine and metabolic cues. Cytokines such as tumor necrosis factor-alpha (TNF-α) and macrophage colony-stimulating factor (M-CSF) in the TME are key inducers of the M2 phenotype[25,29,30]. Concurrently, tumor-derived metabolic products (e.g., lactate and lipid intermediates) activate signaling pathways involving peroxisome proliferator-activated receptor gamma (PPARγ) and hypoxia-inducible factor-1α (HIF-1α), which reinforce M2 polarization[31-34]. For instance, lactate produced by tumor cells can mediate histone lactylation in TAMs, upregulating genes like NUPR1 to enhance their immunosuppressive function[35].
TAMs mediated immune suppression mechanism: factor secretion and signal metabolism regulatory network
Among the immunosuppressive factors secreted by TAMs, IL-10, TGF-β, and the chemokine CCL2 are key molecules that play a central role, forming an “inhibition-enhancement-feedback loop” in the immunosuppressive chain[36,37].
IL-10 directly inhibits the proliferation and cytotoxic function of effector T cells [including CD8+ Cytotoxic T Lymphocytes (CTLs)] and reduces the antigen-presenting efficiency of dendritic cells (DCs), ultimately inducing local immune tolerance in tumors[36]. TGF-β suppresses the activation and function of CD4+ helper T cells and CD8+ effector T cells, and its upregulation of SOX18 promotes Treg accumulation while enhancing tumor cell invasiveness via EMT induction[38,39]. CCL2 recruits peripheral monocytes to replenish the TAM pool and attracts other immunosuppressive cells, forming a positive feedback loop of immune suppression[40,41]. Additionally, M2-like TAMs express programmed death ligand-1 (PD-L1), which binds to programmed death-1 (PD-1) on CD8+ T cells to directly induce T cell exhaustion[42,43].
The secretion of IL-10, TGF-β, and M2 polarization is primarily regulated by classical signaling pathways such as nuclear factor kappa B (NF-κB), signal transducer and activator of transcription 3 (STAT3), and Phosphoinositide 3-Kinase/Protein Kinase B (PI3K/AKT)[44-46]. Recent studies confirm that metabolic reprogramming is a key mechanism sustaining TAM immunosuppressive function, with targeting relevant pathways enhancing immunotherapy effectiveness[33].
MDSCs
MDSCs are a heterogeneous population of immature bone marrow-derived cells with immunosuppressive functions. They originate from myeloid progenitor cells that fail to differentiate normally under pathological conditions such as cancer, expanding as abnormal neutrophils and monocytes[47]. Initially lacking immunosuppressive activity[48], these precursor cells are converted into functionally immunosuppressive MDSCs within TME under the influence of inflammatory and immunosuppressive cytokines including sTNF, TGF-β, IL-10, and IL-1β[49].
Types of MDSCs
MDSCs are primarily divided into two categories: polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs/G-MDSCs) and monocytic myeloid-derived suppressor cells (M-MDSCs), distinguished by their granulocyte or monocyte lineage origin. PMN-MDSCs are phenotypically and morphologically similar to neutrophils, while M-MDSCs resemble monocytes. In mice, they are defined as PMN-MDSCs (CD11b+GR1+Ly6G+Ly6Clo) and M-MDSCs (CD11b+GR1+Ly6G-Ly6Chi)[50,51]. In humans, PMN-MDSCs are HLA-DR-CD11b+CD14-CD15+CD33Mid, and M-MDSCs are HLA-DR-CD11b+CD14+CD15-CD33high, with Lin-HLA-DR-/loCD33+ cells identified as immature eMDSCs[52]. These subpopulations represent different differentiation states within the same lineage.
The immunosuppressive function of MDSCs
MDSCs exert immune evasion through multiple mechanisms, including inhibiting T cell function, inducing Tregs, and suppressing immune responses via ligand-receptor interactions. M-MDSCs accumulate in cirrhotic patients and exhibit stronger T cell suppressive activity[53]. The most common mechanism involves releasing Arg1 and NO to inhibit T cell proliferation, while secreted TGF-β suppresses T cell and NK cell functions. MDSCs recruit and enhance Treg activity through the C-X-C motif chemokine ligand 16 (CXCL16)-C-X-C motif chemokine receptor 6 (CXCR6) axis interaction, with Tregs secreting TGF-β to induce M-MDSC proliferation and immunosuppressive activity[54-56]. MDSCs also upregulate PD-L1 under hypoxic conditions via HIF-1α[57], directly binding to PD-1 on T cells to inhibit activation and promote tumor metastasis and drug resistance.
MDSCs in TIME of HCC
The occurrence of HCC is accompanied by chronic inflammation and liver fibrosis, with MDSC accumulation further promoting an immunosuppressive hepatic microenvironment. E-twenty-six-specific sequence variant 5 (ETV5) promotes S100A9 secretion in liver cancer cells, recruiting and activating MDSCs that secrete inhibitory cytokines and highly express PD-L1 to inhibit cytotoxic CD8+ T cells[58]. SLC7A2 deficiency may upregulate CXCL1 and recruit MDSCs through the PI3K/Akt/NF-κB pathway[59]. In HBV-related HCC, the frequency of CD14+HLA-DR-/low MDSCs is elevated, inducing Tregs and suppressing CD8+ T cells via high arginase activity and the extracellular signal-regulated kinase (ERK)/interleukin-6 (IL-6)/STAT3 pathway[60]. During metabolic dysfunction-associated steatohepatitis (MASH) progression, urokinase-type plasminogen activator receptor (uPAR) expression shifts from hepatic stellate cells (HSCs) in early stages to MDSCs in advanced disease, marking inhibitory TIME establishment[61].
Tregs
Tregs are a subset of T cells characterized by Foxp3, CD25, and CD4 expression, playing a crucial role in immune suppression. They maintain immune tolerance, prevent autoimmune diseases, and regulate the tumor immune environment by suppressing unnecessary immune responses.
Types of Tregs
Thymus-derived Tregs (tTregs) develop in the thymus and acquire self-tolerance through selection, expressing the Foxp3 transcription factor[62]. Peripheral Tregs (pTregs) are induced from peripheral CD4+ T cells in specific microenvironments such as chronic inflammation, infection, or tumor settings[63]. Both subsets suppress effector T cell activity to maintain immune system balance.
The immunosuppressive function of Tregs
Tregs exert immune suppression through secretion of immunosuppressive cytokines, cell contact-dependent mechanisms, and metabolic pathway regulation. TGF-β1 is the predominant immune-related subtype, directly inhibiting effector T cell proliferation and function, and activating tumor cell-derived TGF-β1 via GARP and αvβ8 integrins in TME to inhibit CD8+ T and NK cells and promote M2 macrophage polarization[64]. IL-10 inhibits antigen presentation by macrophages and DCs, amplifies its own secretion via autocrine signaling, and induces myeloid cells to upregulate PD-L1[65]. IL-35 works with IL-10+ Tregs via the STAT3/STAT1/STAT4-BLIMP1 axis to induce CD8+ tumor-infiltrating lymphocytes (TIL) exhaustion[66]. Tregs express Cytotoxic T Lymphocyte-Associated Protein-4 (CTLA-4), which mediates CD80/CD86 consumption, releasing free PD-L1 to bind PD-1 on effector T cells, exerting dual immunosuppressive effects[67]. Lymphocyte activation gene 3 (LAG-3), selectively expressed in inducible Tregs, is crucial for their suppressive activity independent of the Foxp3 pathway[68].
Tregs in TIME of HCC
LTβR downregulates PRDM1 via N-glycosylation to initiate FOXP3 transcription, with HCC cell overexpression of glycolysis-related enzymes hindering LTβR N-glycosylation to promote Foxp3+ Treg infiltration[69]. TGF-β1 upregulates SOX12 via the TGFβR1-Smad2/3/4 pathway and SOX18 via the Smad2/3 complex, enhancing Treg infiltration and immunosuppressive activity while reducing CD8+ T cell function[38,70]. In HBV-related HCC, CCR4+ Tregs are dominant, expressing higher immunosuppressive cytokines and exhibiting stem cell-like characteristics to significantly inhibit CD8+ T cell proliferation and cytotoxicity. Tumor cells secrete CCL22 to recruit CCR4+ Tregs, creating an immunosuppressive environment that induces PD-1 expression on CD8+ T cells and promotes macrophage and MDSC accumulation[71].
Heterogeneity of immunosuppressive cells in HCC
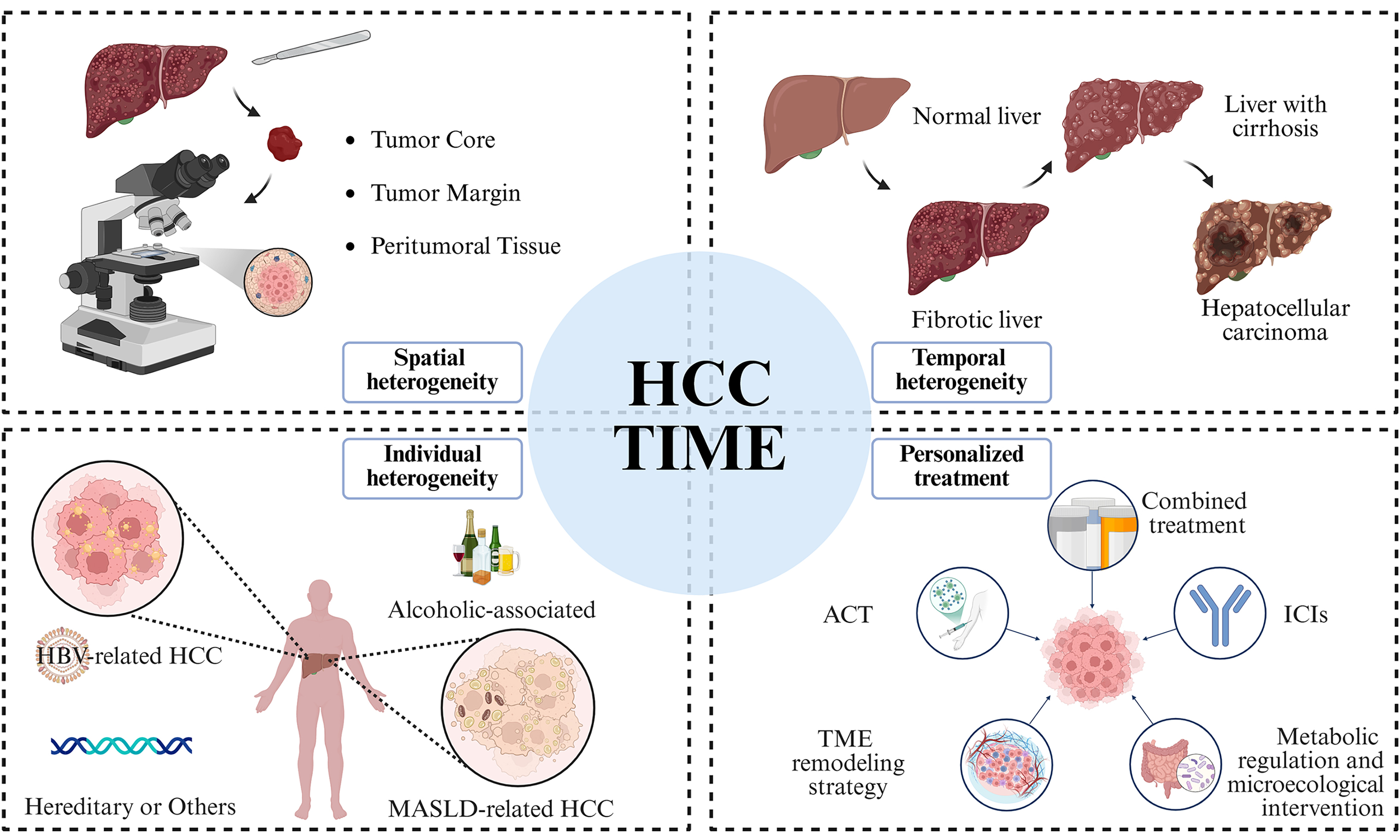
The heterogeneity of M2-like TAMs exhibits distinct etiological and spatial specificity. In HBV-related HCC, the bacteria-dominant intratumoral microbial subtype is associated with higher M2 infiltration and specific metabolic reprogramming[72], while the CK19+ subtype specifically enriches secreted phosphoprotein 1-positive (SPP1+) TAMs[23]. During nonalcoholic fatty liver disease (NAFLD) progression, the NAFLD-mSII subtype linked to high cirrhosis risk shows mixed M1/M2 infiltration[73]. Spatially, M2 macrophages are enriched at the tumor margin, forming a metabolic feedback loop with tumor cells to maintain immunosuppression[74].
MDSC phenotype and function evolve with underlying liver disease. In HBV-related HCC, CD14+HLA-DR-/low MDSCs are elevated, while during MASH progression, uPAR expression shifts to MDSCs in advanced stages[61]. Infiltration of MDSCs and SPP1+ macrophages correlates with poor prognosis via interactions like SPP1-CD44[61]. Tregs are integrated in the immunosuppressive network, with their expansion and activation driven by MDSCs in contexts like HBV-related HCC, collaborating with other cells to weaken anti-tumor immunity[60,75-77].
In summary, M2 macrophages, MDSCs, and Tregs are shaped by HCC etiology and tumor molecular subtypes, forming a heterogeneous immunosuppressive ecosystem [Figure 1]. Deciphering this heterogeneity is essential for understanding immune evasion and developing precision immunotherapies.
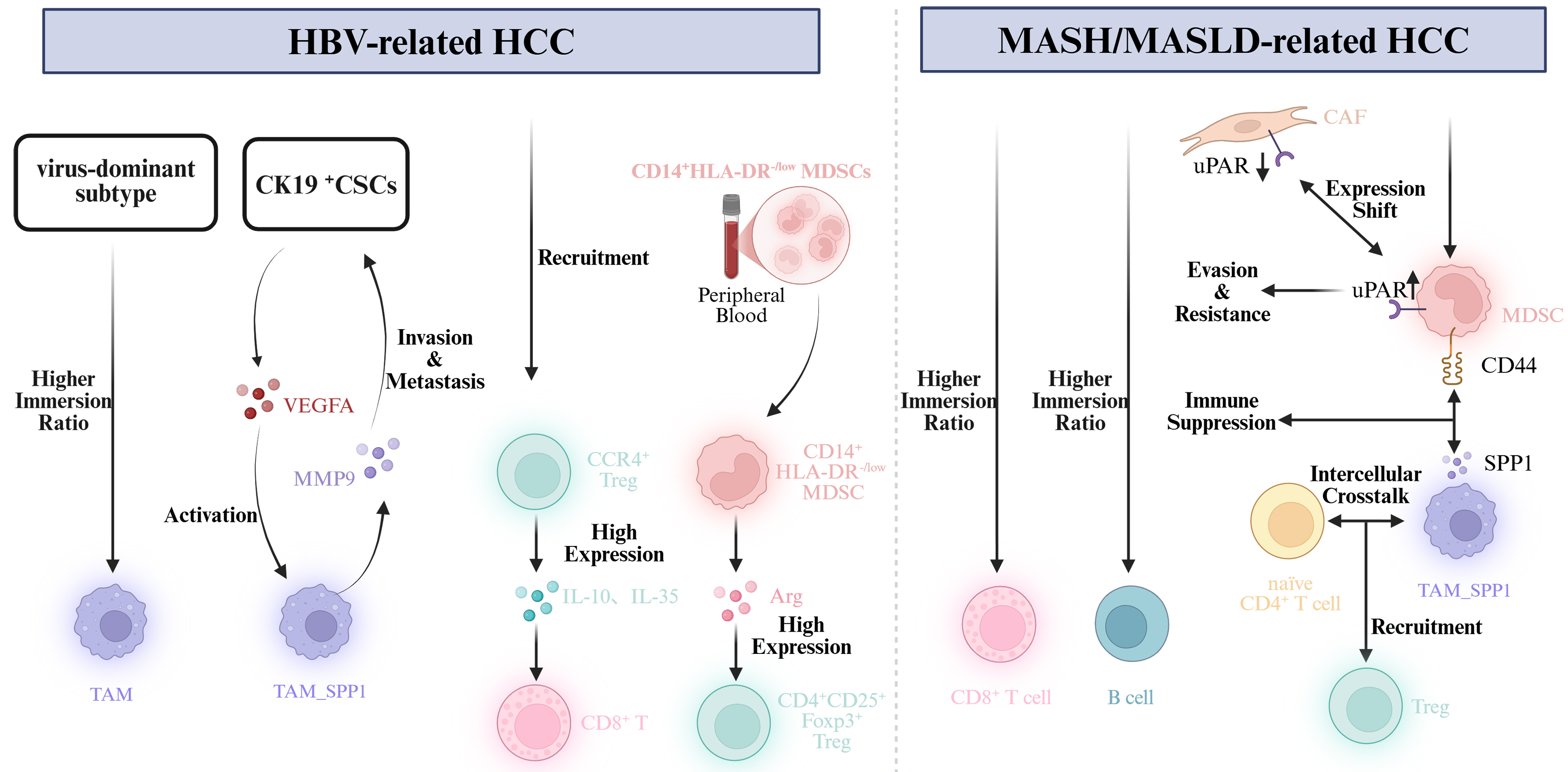
Figure 1. Comparative differences in immune microenvironment characteristics of HCC with different etiologies. Created in BioRender. HBV: Hepatitis B virus; HCC: hepatocellular carcinoma; CK19: cytokeratin 19; CSCs: cancer stem cells; VEGFA: vascular endothelial growth factor A; TAM: tumor-associated macrophage; MMP9: matrix metalloproteinase 9; CCR4: C-C chemokine receptor type 4; Treg: regulatory T cell; SPP1: secreted phosphoprotein 1; HLA-DR: human leukocyte antigen-DR isotype; MDSC: myeloid-derived suppressor cell; Arg: arginase; Foxp3: forkhead box P3; uPAR: urokinase-type plasminogen activator receptor; MASH: metabolic dysfunction-associated steatohepatitis; MASLD: metabolic dysfunction-associated steatotic liver disease.
FUNCTIONAL IMMUNE CELLS AND ANTI-TUMOR IMMUNITY
In the TIME, the functional activity of immune cells directly impacts anti-tumor immunity. Functional immune cells such as CD8+ CTLs, CD4+ helper T cells, B cells, NK cells, and DCs play key roles in immune surveillance, anti-tumor responses, and immune tolerance[71,78,79].
CTLs: antigen recognition, functional exhaustion, and recovery
CD8+ CTLs are key anti-tumor effector cells in HCC TIME, exerting cytotoxic effects by recognizing tumor antigens presented via major histocompatibility complex-I (MHC-I) molecules. Their functional status directly affects tumor progression and immunotherapy efficacy.
Tumor cells reduce MHC-I expression via lysosomal degradation, weakening antigen presentation and inhibiting CTL recognition[80]. Tumor-associated neutrophils (TANs) highly express PD-L1, inhibiting CD8+ T cell activation and cytotoxicity while promoting exhaustion[81]. High tumor metabolism causes nutrient depletion, leading to CTL functional exhaustion with reduced secretion of cytotoxic molecules and anti-tumor cytokines[82]. LCFAs accumulation induces mitochondrial dysfunction in CD8+ T cells, suppressing fatty acid catabolism and function[82]. Additionally, tumor cell secretion of immunosuppressive factors, recruitment of inhibitory cells, and high PD-L1 expression contribute to CTL exhaustion via PD-1/PD-L1 pathway activation[83].
Stem cell-like CD8+ T cells (TCF1+ TIM3- CD28+) exhibit strong proliferative potential, while terminally differentiated cells (TCF1- TIM3+ PD-1+) lack proliferation ability and show typical exhaustion characteristics[84]. Functional reprogramming is coordinated by the JAK-STAT pathway, with IL-2/IL-15 activating STAT5, IL-21 activating STAT3, and IL-6 activating STAT3/STAT1 heterodimers. In chronic hepatitis B patients, STAT3 phosphorylation correlates with CTL functional recovery[85,86].
CD4+ helper T cell subsets: cytokine network
Different CD4+ helper T cell subsets affect tumor immunity via specific cytokines, with Th1 and Th17 playing complex roles in HCC. Th1 cells secrete Interferon-γ (IFN-γ), IL-2, and TNF-α to promote CD8+ T and NK cell activation and enhance anti-tumor immunity. Their differentiation depends on IFN-γ-STAT1-T-bet signaling, with T-bet regulating chemokine expression and migration[87]. IFN-γ activates macrophages and enhances DC antigen presentation. Probiotic BLE may enhance liver Th1 function by upregulating T-bet and promoting cytokine secretion[88]. Th17 cells secrete IL-17, activating NF-κB and STAT3 pathways to promote tumor development[87]. Differentiated from naïve CD4+ T cells under IL-6 and TGF-β1 induction, Th17 cells are regulated by retinoic acid-related orphan nuclear receptor γt (RORγt), with IL-23 enhancing RORγt expression via STAT3 to promote expansion and stabilization[89-91].
B cells and TABs: anti-tumor vs. immunosuppression
B cells play a dual role in TIME, both promoting anti-tumor immunity and mediating suppression. Tumor-infiltrating B cells (TIL-Bs) and tumor-associated B cells (TABs) exhibit significant heterogeneity. B cell presence in liver cancer correlates with improved patient survival, with activated subsets enhancing CD8+ T cell activation[92]. μMT mice lacking B cells show aggravated HCC progression and impaired CD8+ T cell activation[92]. B cells participate in tertiary lymphoid structures (TLS) formation, promoting local immune memory and effector cell generation[93,94].
B cells also mediate immune suppression by secreting IL-10 or expressing PD-L1. In HCC, TABs are recruited by TAMs via the CXCL12/CXCR4 axis, expressing PD-L1 to inhibit immune effector cells[95]. B cell-derived GABA reprograms macrophages to induce an immunosuppressive environment[96]. IgG+ plasma cells promote tumor-promoting macrophage formation in HCC, while IgA+ plasma cells induce MDSC activation in colorectal cancer liver metastasis[97]. B cell abundance and activation status predict survival and immunotherapy response, with high infiltration indicating better prognosis and stronger anti-tumor immunity[98,99].
NK cells: suppressed by NKG2A and TIGIT signaling mechanisms
NK cell activity is regulated by activating and inhibitory receptors, with natural killer group 2A (NKG2A) and T Cell Immunoreceptor with Ig and ITIM Domains (TIGIT) pathways crucial for function inhibition. In HCC patients, NKG2A upregulation reduces NK cell anti-tumor effects, while blocking NKG2A restores activity and enhances antibody-dependent cellular cytotoxicity (ADCC)[100]. In HBV-related HCC, TIGIT+ TIM-3+ NK cells exhibit exhaustion with weakened killing function, reduced cytokine secretion, and impaired proliferation[101]. Fibrinogen-like protein 1 (FGL1) inhibits CD8+ T and NK cells via LAG-3 binding, promoting HCC progression[102]. LCACs accumulation induces iNKT cell senescence, weakening immune surveillance[103]. Enhanced STAT3 activity and PRDM10 downregulation in liver NK cells contribute to dysfunction and exhaustion[104,105]. Soluble MHC Class I Chain-Related A (sMICA) released by tumor cells disrupts NKG2D signaling, impairing NK cell activation[106]. These mechanisms collectively lead to NK cell exhaustion and immune evasion.
DCs and antigen presentation defects
DCs are key antigen-presenting cells, cross-presenting tumor antigens to activate naïve T cells and induce anti-tumor immunity. After capturing antigens, DCs present them to CD8+ T cells via MHC-I molecules, promoting CTL differentiation and tumor cell killing[81,107]. In TME, DCs often exhibit antigen-presenting defects. In HCC, DC dysfunction is characterized by reduced maturity, decreased CD80/CD86 expression, and reduced IL-12 secretion, preventing effective T cell activation and exacerbating immune tolerance[108,109]. The JAK2-STAT3 pathway activation negatively impacts DC maturation and function[109]. Tumor-associated exosomes (TAEs) alter DC cytokine secretion, reducing IL-12 and increasing IL-10 and TGF-β to impair immune activation[110,111].
Heterogeneity of functional immune cells in HCC
CD8+ T cell infiltration and function are pivotal in HCC. HBV-related HCC shows higher infiltration and tumor reactivity, while metabolic dysfunction-associated steatotic liver disease (MASLD)-related HCC exhibits T cell dysfunction due to tumor glucose consumption via NSUN2-mediated metabolic reprogramming[112,113]. HBV/MASLD co-existing HCC has elevated precursor-exhausted CD8+ T cells, associated with better immunotherapy responses[112], while Wnt-non-mutant metastatic sites show terminal CD8+ T cell exhaustion mediated by immunosuppressive B cells[114]. In alcohol-associated HCC, reduced CD69- CD4+ T cell frequency with elevated PD-L1 suggests impaired immune surveillance[115]. In MASH-HCC, naïve CD4+ T cell crosstalk with SPP1+ macrophages via macrophage migration inhibitory factor (MIF) signaling promotes Treg infiltration[116]. In MASLD-driven HCC, activated B cell subsets correlate with favorable prognosis, co-localizing with CD45RO+ CD8+ T cells in TLS[92,117]. However, in Wnt-non-mutant metastatic microenvironments, B cells adopt an immunosuppressive phenotype to drive CD8+ T cell exhaustion[114]. HBV-related HCC shows increased “adaptive NK cells” with limited anti-tumor activity[118,119]. In MASH-HCC, tumor cell metabolism-associated genes (e.g., ACSL4) modulate NK cell-mediated clearance via ferroptosis regulation[120]. DC heterogeneity in HCC is still insufficient. In summary, immune cell abundance, function, and spatial distribution are regulated by etiology, tumor genetics, and local signaling networks, shaping TIME characteristics and treatment responses.
THE INTERACTION NETWORK BETWEEN INHIBITORY CELLS AND FUNCTIONAL CELLS
The HCC TIME operates as an integrated network where stromal, immune, and tumor cells mutually reinforce immunosuppression. CAFs and HSCs act as primary architects, driving monocyte differentiation into MDSCs via IL-6/STAT3 and SDF-1/CXCR4 signaling[121]. Beyond cytokines, the physical environment - specifically Neutrophil Extracellular Traps (NETs) - induces TLR4-mediated mitochondrial oxidative phosphorylation to reshape myeloid metabolic profiles[33,122].
This network is sustained by “amplification loops”: MDSCs secrete CCL2 to replenish the TAM pool, while lipid-laden TAMs recruit and differentiate Tregs via the CCL20/CCR6 and S1P axes[40,78]. Unique TAM subsets further refine this niche; TREM2+ TAMs modulate CXCL9/Galectin-1 expression, while FABP5+ and XOR-deficient TAMs drive suppressive metabolite accumulation[123]. Non-classical regulators also play vital roles: Bregs impair CD8+ T-cell function via IL-21R-STAT1 or PD-L1 expression, and IgA+ B cells promote fibrosis[124,125]. In HBV-related HCC, B cells communicate with CAFs through the pleiotrophin (PTN)-syndecan-1 (SDC1)/nucleolin (NCL) axis to accelerate progression[126]. Meanwhile, endothelial cells secrete CXCL12 to recruit MDSCs and directly arrest the differentiation of naïve CD8+ T cells, creating an immunosuppressive vascular barrier[127].
Ultimately, this crosstalk facilitates evasion by targeting both innate and adaptive killers. Beyond exhaustion, MDSCs suppress NK cells via NKp30 binding, while tumor-derived TGF-β flips NK cells into dysfunctional group 1 innate lymphoid cells (ILC1)-like phenotypes[121]. Specific HCC markers like EpCAM and CD155 further confer resistance to cytotoxicity. Moreover, high-lactate and high-lipid conditions expand regulatory DCs, impairing essential DC-T cell crosstalk[128,129]. These interactions are highly etiology-specific: In NAFLD/MASH, lipid dysfunction and MATR3 deficiency accelerate CD4+ T cell apoptosis, whereas HBV-related HCC establishes an “exhaustion hub” via itaconate and SOX18 signaling to resist NK-cell-mediated lysis[33,38,127,130].
HETEROGENEITY OF TIME IN HCC: AN INTEGRATED THREE-DIMENSIONAL PERSPECTIVE
The preceding sections have delineated the core cellular components of the HCC TIME and their intricate intercellular crosstalk. However, these elements do not exist in a static or uniform state; rather, they exhibit profound heterogeneity that acts as a primary driver of immune evasion and a fundamental determinant of variable treatment responses. To systematically decode this complexity, we map these dynamic variations onto our proposed Integrated Three-Dimensional Heterogeneity Model. The following discussion will elucidate how this heterogeneity manifests and is mechanistically regulated across three interconnected axes: variations among individual patients driven by distinct pathogenic backgrounds (Etiology), localized compartmentalization across discrete tumor niches (Spatial Topology), and phased immunological shifts throughout disease progression (Temporal Evolution).
Heterogeneity among individual patients
The heterogeneity of the immune microenvironment across individual patients is likely a key determinant of variable responses to immunotherapy. Patients exhibit inherent differences in their immune systems, reflected in baseline variations in immune responsiveness, immune evasion mechanisms, and immune tolerance.
Beyond these intrinsic factors, the immunological profile of liver cancer patients is shaped by a combination of genetic, environmental, and lifestyle influences. Underlying liver disease etiologies - such as hepatitis virus (HBV or HCV) infection, alcoholic liver disease, and nonalcoholic fatty liver disease - further remodel the immune landscape in which the tumor develops[131]. The main heterogeneity of immune features in HCC caused by different etiologies is summarized in Table 1.
Immune features, tumor phenotypes and therapeutic responses of HCC by etiological subtypes
| Etiological subtype | Core immune features | Tumor phenotype | Therapeutic response characteristics | Ref. |
| HBV-related HCC | Dysfunctional/exhausted HBV-specific T cells; Increased Tregs infiltration | “Warm” tumor (partial immune infiltration) | Moderate response to ICIs; Dependent on functional T cell activation | [112,132] |
| MASLD-related HCC | Abundant CD8+ T cell infiltration; Enhanced tumor vascular normalization | “Cold-to-warm” transition phenotype | Variable ICI efficacy; Responsive to metabolic intervention | [112,133,134] |
| HBV+MASLD+ coinfected HCC | Enriched precursor-exhausted CD8+ T cells; Optimized immune cell crosstalk | Immune-responsive phenotype (“Hot” tumor) | Immunotherapy offers the greatest benefits. | [112,134] |
| Non-HBV/non-MASLD-related HCC | Weak antigen-specific T cell response; Balanced inhibitory/effector immune cell ratio | Heterogeneous phenotype (A predominantly “cold” tumor microenvironment) | Intermediate ICI response | [112,135] |
In addition to these contextual factors, differential response rates to immunotherapy in HCC are closely linked to the composition and functional states of specific immune cell populations within the TME. The activity, abundance, and immunosuppressive factor secretion of various cell types - including Tregs, NK cells, DCs, MDSCs, TAMs and CAFs - collectively influence the capacity for immune evasion and ultimately shape the efficacy of immunotherapeutic interventions[7,60,76,136].
Intratumoral heterogeneity
HCC is characterized by pronounced local progression. Different tumor regions - such as the core, invasive margin, and peritumoral area - display marked variations in immune cell infiltration, metabolic profiles, and mechanisms of immune evasion.
Spatial heterogeneity: tumor core vs. margin vs. peritumoral tissue
The infiltration and function of immune cells within HCC exhibit substantial spatial heterogeneity across different tumor regions [Figure 2]. The tumor core is typically characterized by a
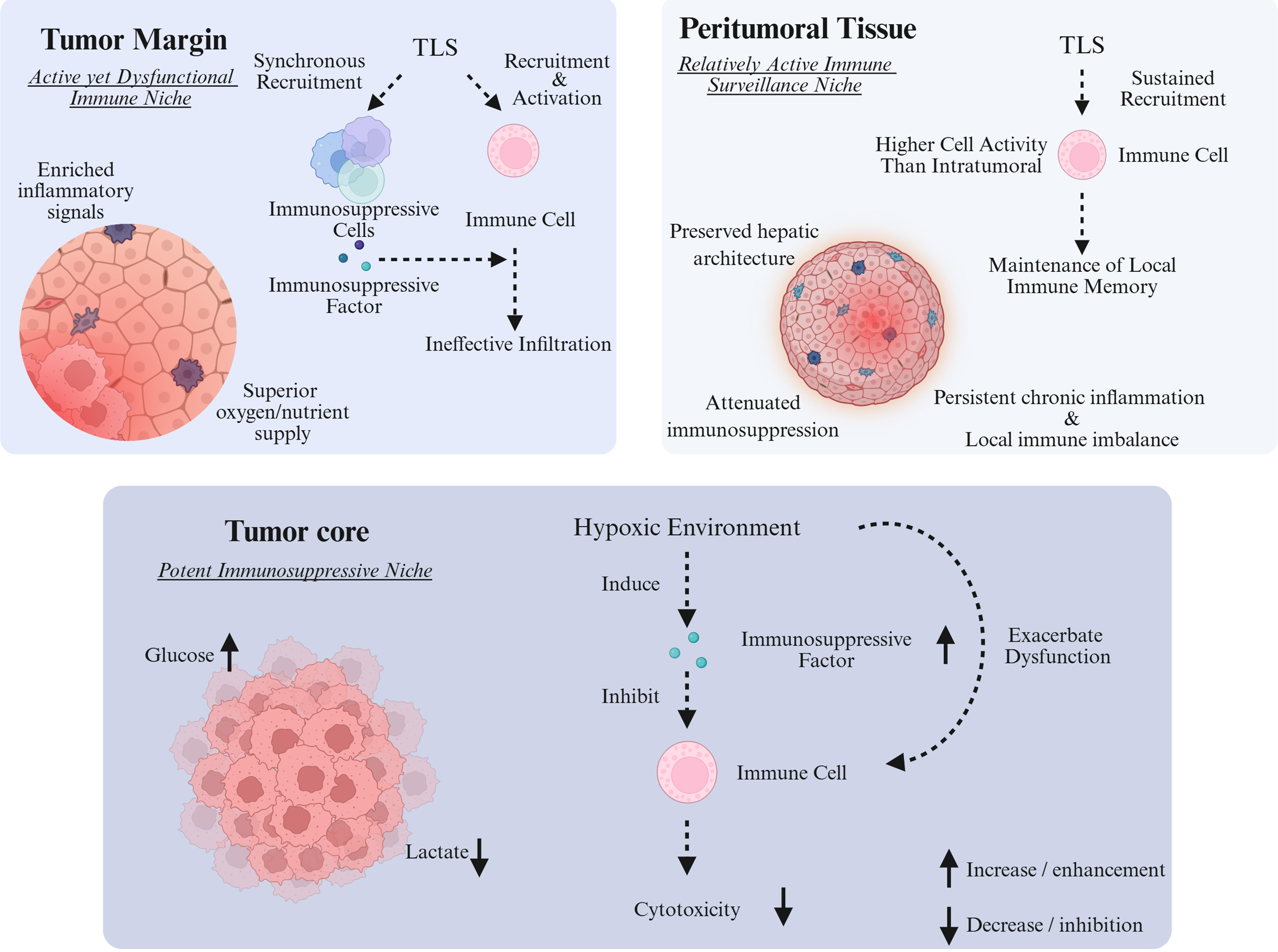
Figure 2. Spatial heterogeneity of the immune microenvironment in different local regions of HCC. Created in BioRender. HCC: Hepatocellular carcinoma; TLS: tertiary lymphoid structure.
By contrast, the tumor margin and peritumoral tissues generally display more active immune engagement. These regions often show increased infiltration of immune cells. Studies have shown that peritumoral tertiary lymphoid structures (pTLS) help recruit B cells, T cells (including CD3+, CD8+, and CD20+ subsets), and DCs to the invasive margin and surrounding stroma. Furthermore, NKs in the peritumoral compartment tend to exhibit stronger antitumor activity compared to their intratumoral
Nevertheless, immune function in these areas is frequently modulated by tumor-associated immunosuppressive cells - such as Tregs and MDSCs - which create a locally imbalanced immune milieu[142,143]. Consequently, although immune cell density is higher at the tumor margin and in peritumoral tissue, their effector functions are often restrained, enabling the tumor to evade immune surveillance.
Temporal heterogeneity: phased manifestations in the immune evolution of HCC
The temporal heterogeneity of immune evolution in HCC unfolds across the continuum from cirrhosis (a pre-neoplastic stage) through early HCC to advanced disease [Figure 3]. This progression exhibits a bidirectional shift, characterized by an “increasing gradient of immune suppression” alongside a “decreasing gradient of anti-tumor immunity”.
Figure 3. Dynamic evolution of immune status across stages of HCC progression. Created in BioRender. HCC: Hepatocellular carcinoma; Treg: regulatory T cell; MDSC: myeloid-derived suppressor cell; TNF: tumor necrosis factor; TGF-β: transforming growth factor-beta; Arg-1: arginase-1; MMPs: matrix metalloproteinases; CXCL12: C-X-C chemokine ligand 12; NO: nitric oxide; ROS: reactive oxygen species.
During the cirrhosis stage, the immune microenvironment is characterized by chronic inflammation that drives the establishment of immune tolerance, thereby laying the groundwork for an immune-permissive milieu conducive to liver cancer development. The function of innate immune cells becomes dysregulated: KCs shift from an anti-tumor M1 phenotype toward a pro-inflammatory, pro-fibrotic M2-like state. These cells sustain chronic inflammation through the secretion of TNF and IL-6, while simultaneously inhibiting T-cell activation via TGF-β. HSCs, activated by inflammatory signals, differentiate into myofibroblasts that not only promote fibrosis through collagen deposition but also foster the accumulation of Tregs by expressing PD-L1 and secreting IL-10[144]. Adaptive immunity undergoes selective attrition. In metabolic-associated cirrhosis, such as non-alcoholic steatohepatitis (NASH), linoleic acid-mediated lipotoxicity leads to a pronounced loss of CD4+ T cells. Although CD8+ T cells are not significantly depleted, they enter a “pre-exhausted” state marked by diminished IFN-γ secretion and cytotoxicity[130]. At the molecular level, the immune network exhibits a procancer imbalance, with a skewed ratio of proinflammatory cytokines (e.g., TNF, IL-6) to antiinflammatory factors (e.g., IL-10, TGF-β). Chronic inflammation continuously induces DNA damage and the accumulation of pre-mutagenic alterations in hepatocytes, while anti-inflammatory mediators suppress immune-mediated clearance of mutated cells. This results in a paradoxical state of “inflammation-driven carcinogenesis coupled with immune tolerance”, enabling the emergence and persistence of potentially malignant clones[145].
In early-stage HCC, where tumor burden remains relatively low, the immune landscape displays a precarious equilibrium between local immunosuppression and anti-tumor immunity. Anti-tumor immune cells retain partial functionality: liver-resident NK cells (particularly the CD49a+DX5- subset) and a subset of non-exhausted CD8+ T cells exert cytotoxic effects via perforin and granzyme B secretion. Although DC numbers are reduced, they can still present tumor antigens in limited quantities, sustaining a weak but detectable anti-tumor response[146]. Concurrently, immunosuppressive cells begin to accumulate locally. Tregs and MDSCs are selectively enriched at the tumor margin, where they inhibit CD8+ T cell and NK cell function through IL-10 secretion and arginase release. TAMs further polarize toward an M2-like phenotype, promoting early angiogenesis via VEGF secretion. Immune structures and molecular profiles undergo early remodeling. Intratumoral tertiary lymphoid structures (iTLS) - composed of B cells, T cells, and DCs - form in approximately 30% of early HCC cases and can locally activate adaptive immunity, temporarily restraining tumor progression. Meanwhile, HCC cells initiate early immune evasion, though expression of immune checkpoint molecules such as PD-L1 and CTLA-4 remains low and not yet sufficient to fully block anti-tumor immunity[145,147].
In advanced HCC, a marked increase in tumor burden drives the formation of a multidimensional immunosuppressive network and highly evolved immune evasion mechanisms. Adaptive immune cells become profoundly exhausted. CD8+ T cells enter a terminally exhausted state, characterized by high coexpression of immune checkpoint molecules (PD-1, TIM-3, LAG-3), loss of proliferative capacity and cytotoxicity, and inability to secrete IFN-γ or granzymes. CD4+ T cell numbers decline further, with Tregs expanding to constitute over 40% of the remaining pool. Tregs enhance suppression by sequestering IL-2 and inhibiting dendritic cell maturation[148]. Innate immune function is also severely paralyzed. NK cells are diminished due to IL-15 deprivation and MDSC-derived reactive oxygen species (ROS), losing their cytotoxic capacity. MDSCs, particularly the granulocytic subset, accumulate extensively and suppress CD8+ T and NK cells by depleting arginine and releasing nitric oxide (NO) and ROS. TAMs fully polarize into a pro-metastatic M2-like phenotype, secreting matrix metalloproteinases (MMPs) to promote invasion, CXCL12 to recruit tumor cells, and expressing PD-L1 to directly inhibit T-cell activation[149]. A self-reinforcing “vicious cycle” dominates TIME. Tumor cells enhance glycolysis, depleting local glucose and starving CD8+ T cells of energy. Concurrently, lactate secreted by tumor cells and TAMs acidifies the microenvironment, further impairing Tcell function and promoting MDSC enrichment. High expression of immune checkpoint molecules (PD-L1, CTLA-4) on both tumor and suppressive immune cells establishes multiple, overlapping layers of immune blockade[148,149].
THERAPEUTIC STRATEGY TARGETING TIME OF HCC
We learned that the etiology heterogeneity of HCC is closely related to immune cells heterogeneity, and this complex relationship will have a significant impact on therapeutic response variation [Figure 4]. Given the heterogeneous features of the TIME of HCC - such as inter-patient differences in immune cell composition and microenvironmental variations between the tumor core and margin - single-target therapies are inadequate to cover all contexts. Overcoming the therapeutic resistance imposed by this heterogeneity requires a multidimensional targeting strategy. Recent years have witnessed significant advances in therapeutic strategies targeting the TIME of HCC, notably in immunotherapy, exemplified by ICIs.
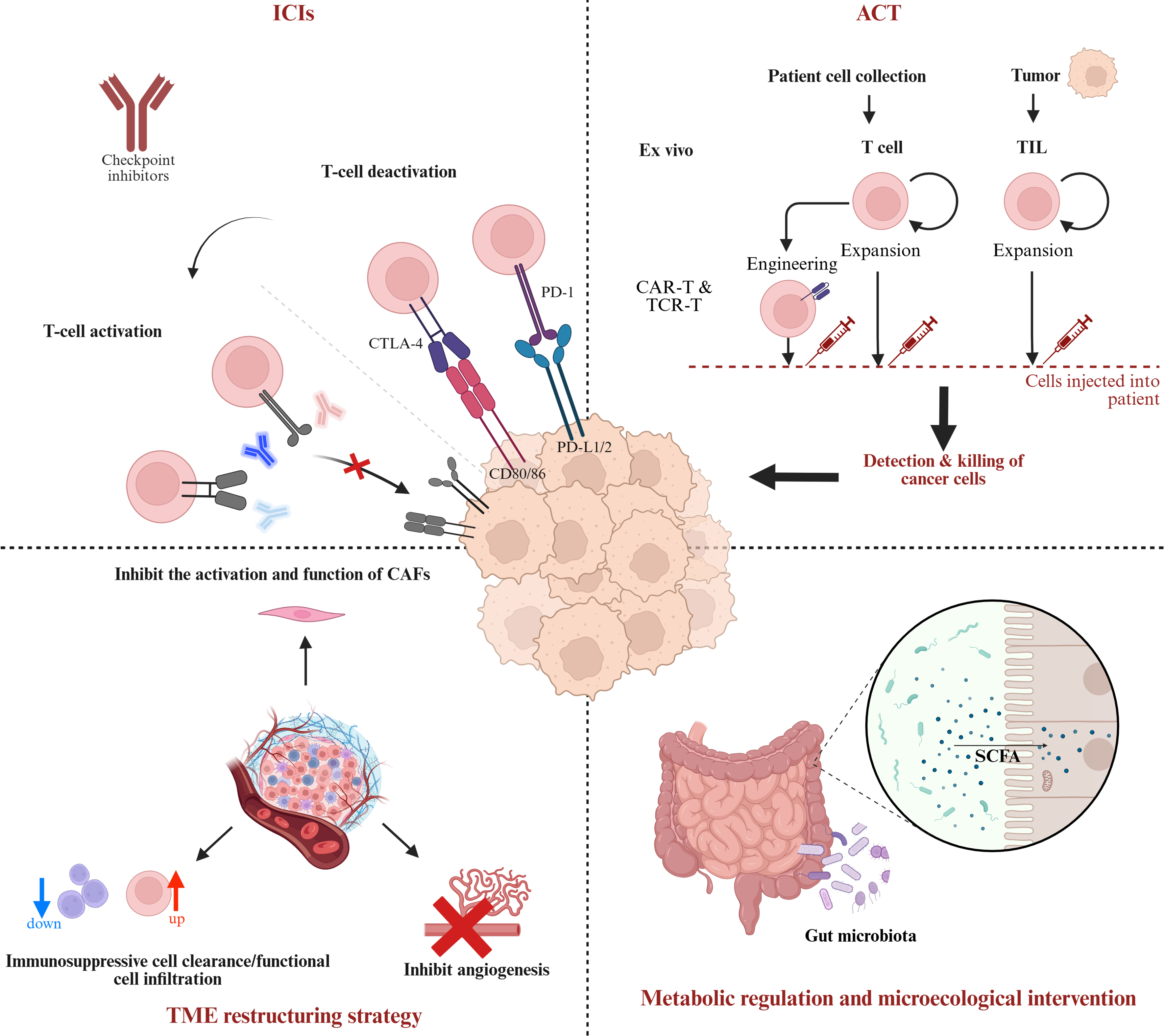
Figure 4. Multidimensional regulatory strategies for HCC immunotherapy and tumor. Created in BioRender. ICI: Immune-checkpoint inhibitor; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; PD-1: programmed cell death protein 1; PD-L1: programmed cell death ligand 1; TIL: tumor-infiltrating lymphocyte; CAR-T: chimeric antigen receptor T cell; TCR-T: T-cell receptor-engineered T cell; TME: tumor microenvironment; ACT: adoptive cell therapy; SCFA: short-chain fatty acid; CAFs: cancer-associated fibroblasts.
ICIs
ICIs have emerged as a pivotal tool in HCC immunotherapy, achieving substantial clinical progress and becoming an indispensable treatment strategy. ICIs function primarily by blocking negative regulatory signals between tumor cells and immune cells, thereby restoring T cell-mediated anti-tumor activity and reversing tumor immune evasion. The most extensively utilized ICI targets in clinical practice include PD-1 and its ligand PD-L1, as well as CTLA-4. Additionally, emerging targets such as LAG-3, TIGIT, TIM-3, V-Domain Ig Suppressor of T Cell Activation (VISTA), and Siglec-9 are garnering increasing research interest.
Monoclonal antibodies targeting the PD-1/PD-L1 pathway, such as the anti-PD-1 agents nivolumab and pembrolizumab and the anti-PD-L1 agent atezolizumab, have demonstrated therapeutic efficacy in advanced HCC across multiple clinical trials. The IMbrave150 trial established atezolizumab combined with the anti-angiogenic bevacizumab as a first-line standard, significantly improving overall survival (OS) and progression-free survival (PFS)[150]. Single-cell sequencing data suggest differential benefits across molecular subgroups of advanced HCC with this regimen. Real-world evidence, such as from the AB-real study (n = 296), reported a median OS of 15.7 months, a median PFS of 6.9 months, and an objective response rate (ORR) of 30.8%[151].
Additionally, the combination of nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) has shown significant clinical activity with durable responses, particularly in sorafenib-pretreated patients. In the second-line setting, this combination achieves an ORR of approximately 30% and a median OS of 22 months, demonstrating superior anti-tumor efficacy[151].
CTLA-4 is another key immune checkpoint, which primarily mediates immunosuppression by regulating the activation of naïve T cells. Anti-CTLA-4 monoclonal antibodies, such as tremelimumab and ipilimumab, have demonstrated efficacy in HCC treatment. Their combination with PD-1/PD-L1 inhibitors can yield synergistic effects, enhancing the overall response rate. Emerging immune checkpoint targets, including LAG-3, TIGIT, and VISTA, have attracted growing interest[151,152]. These molecules are involved in modulating T cell activity and maintaining immune tolerance. Preclinical and early clinical studies suggest that inhibitors targeting these pathways may help overcome resistance to PD-1/PD-L1 blockade, whether used alone or combined with anti-CTLA-4 therapy, offering potential for new advances in HCC immunotherapy.
Although ICIs have shown considerable promise in HCC, only a subset of patients achieve a meaningful response, while challenges like therapeutic resistance and immune-related adverse events (irAEs) remain common. The complex TIME of HCC, rich in immunosuppressive cells and signaling pathways, facilitates immune evasion and contributes to low ICI response rates. Additional factors, including tumor heterogeneity, loss of tumor antigen expression, and impaired antigen presentation, further limit efficacy. Consequently, significant research efforts are focused on developing ICI-based combination strategies to overcome resistance and improve outcomes. These include combinations with anti-angiogenic tyrosine kinase inhibitors (TKIs), other targeted molecular therapies, local-regional treatments [e.g., Transarterial Chemoembolization (TACE)], and multi-checkpoint inhibition[153-156].
The advancement of precision immunotherapy in HCC will increasingly depend on biomarker screening to identify responsive patients. Studies suggest that potential predictive markers for ICI efficacy include the density of tumor-infiltrating CD8+ T cells, expression of immune-related genes, serum levels of inflammatory cytokines (e.g., IL-8), and liver function-related indicators [e.g., alpha-fetoprotein (AFP), neutrophil-to-lymphocyte ratio (NLR)][157-159]. Furthermore, a deeper understanding of tumor genomics and the TIME will help explain the differential ICI responses between immunologically “hot” and “cold” tumors, thereby providing a foundation for individualized treatment strategies[160,161].
TME remodeling strategy
Tregs are highly enriched in HCC. Notably, PD-1-expressing Treg subsets demonstrate potent immunosuppressive activity within both the tumor and peripheral blood, effectively hindering effector T cell activation and anti-tumor immunity[162]. Key pathways governing Treg function include immune checkpoints such as PD-1/PD-L1 and LAG-3, whose blockade can attenuate Treg-mediated suppression. Furthermore, Treg recruitment into the TME is critically regulated by chemokine signaling. For example, the CXCL12/CXCR4 axis promotes Treg infiltration in HCC, and its inhibition can reduce Treg accumulation and alleviate local immunosuppression[38,55].
MDSCs, a major class of immunosuppressive cells in the TIME of HCC, promote tumor immune evasion by secreting factors that impair cytokine production and effector T cell function[121]. The metabolic characteristics and signaling pathways underpinning MDSCs and TAMs are being elucidated. For instance, in liver metastasis, they exhibit upregulated CD36-mediated lipid metabolism, which enhances fatty acid uptake and augments their immunosuppressive activity. Targeting CD36 can thereby suppress the tumor-promoting functions of them[163,164].
Current strategies to deplete or inhibit Tregs and MDSCs are inherently synergistic. For example, ICIs such as anti-CTLA-4 antibodies can restore effector T cell function while simultaneously suppressing Treg activity[67]. Furthermore, targeting chemokine receptors like CXCR4 - which is overexpressed in HCC and TME cells - holds promise. In liver cancer, a “dual-hit therapy” using CXCR4 antagonists can block the recruitment of both Tregs and MDSCs to the tumor, thereby alleviating immunosuppression within the TME[55].
Nanodrug delivery systems enable the targeted delivery of siRNA or small-molecule inhibitors. For example, silencing key regulators such as protein tyrosine phosphatase non-receptor type 2 (PTPN2) can promote the polarization of TAMs toward an M1 phenotype and enhance tumor cell immunogenicity[165,166]. Moreover, small molecules like vitamin B3 can modulate the GPR109A signaling pathway, inhibiting the immunosuppressive polarization of myeloid cells while boosting the cytotoxicity of CD8+ T cells[167].
The integration of nanotechnology with radiotherapy, chemotherapy, or targeted therapy can further elevate oxidative stress within the TME. This enhances immune cell infiltration and reduces the recruitment of immunosuppressive cells, creating a synergistic effect that ultimately improves the efficacy of immunotherapy[168-171]. These strategies extend beyond directly targeting immunosuppressive cells; they also remodel the tumor immune landscape by modulating metabolic reprogramming, cellular polarization, and intercellular communication. This multifaceted approach can effectively convert immunologically “cold” tumors into “hot” ones, thereby improving treatment responses in patients with HCC.
Adoptive cell therapy (ACT)
Chimeric antigen receptor T cell (CAR-T) cell therapy is under active investigation for HCC, leveraging its high specificity and potent cytotoxicity[172]. Tumor-associated antigens such as AFP and glypican-3 (GPC3) serve as key targets. AFP-targeted CAR-T cells demonstrate selective cytotoxicity against HCC cells[173]. Furthermore, GPC3-directed CAR-T therapies have progressed to clinical trials, showing favorable safety and preliminary antitumor activity[174-176].
Combining AFP- and GPC3-targeted CAR-T cells, or engineering T cells to secrete bispecific T-cell engagers (BiTEs), may enhance anti-tumor immune responses[177]. However, CAR-T therapy in solid tumors such as HCC remains challenged by the immunosuppressive TME, insufficient T cell infiltration, and tumor antigen heterogeneity, which collectively promote immune evasion and limit therapeutic efficacy[174,178,179].
TIL therapy involves isolating lymphocytes from a patient’s tumor, expanding them ex vivo, and reinfusing them to enhance anti-tumor immunity. This approach shows particular promise in HCC patients with “hot”, immune-infiltrated tumors, which provide a supportive microenvironment for TIL activity. However, its efficacy in HCC is limited by several factors. The TIME, rich in Tregs and MDSCs, can inhibit reinfused TILs. Furthermore, TIL manufacturing is complex, labor-intensive, and exhibits considerable interpatient variability. These challenges - coupled with the need for personalized protocols - currently restrict the broader clinical application of TIL therapy for HCC[180-182].
NK cell therapy is increasingly explored for HCC due to its role in anti-tumor immunity. NK cells can recognize and eliminate MHC-I-deficient tumor cells independently of antigen presentation. Specialized subsets such as γδT cells also hold potential for liver cancer immunotherapy, and modulating NK cell function has been shown to enhance anti-tumor responses[183]. However, NK cell therapy faces challenges within the inhibitory TIME. A key limitation is the lack of sustained proliferative signals after infusion, which curtails long-term efficacy. Strategies such as cytokine engineering or lymphodepletion prior to infusion are being investigated to prolong NK cell persistence and activity, thereby overcoming the suppressive microenvironment and maximizing therapeutic potential[184,185].
Cellular therapies for HCC remain in early clinical development, facing several key challenges including the inhibitory TIME, antigen heterogeneity and loss, manufacturing complexity, and treatment-related toxicity. Looking forward, a novel approach involves nanoscale lipid vesicles (30-150 nm) derived from CAR-T, CAR-NK, or CAR-M cells, termed CAR-Exosomes. These particles inherit the tumor-targeting specificity of their parental cells while leveraging the intrinsic advantages of exosomes. By combining these strengths, CAR-Exosomes represent a promising strategy to overcome current limitations and may provide a breakthrough in next-generation cancer immunotherapy[186,187].
In summary, adoptive cell therapies - such as CAR-T, TIL, and NK cell-based treatments - represent promising therapeutic approaches for HCC[188]. However, their efficacy is limited by the inhibitory TIME and significant tumor heterogeneity. Advances in cellular engineering, rational combination with immunomodulatory agents, and active remodeling of the TIME are therefore critical to enhancing the safety and efficacy of these therapies and improving patient outcomes.
Metabolic regulation and microecological intervention
Lactate and its derived lactylation modifications significantly reshape the immune landscape within TIME. Lactate drives the polarization of TAMs toward an immunosuppressive M2 phenotype while impairing the effector functions of CD8+ T cells, thereby suppressing immune surveillance[189-191]. Furthermore, lactate can activate signaling pathways that upregulate immune checkpoint expression, further dampening anti-tumor immunity.
Interventions targeting lactate metabolism - such as inhibiting lactate dehydrogenase (LDH) or modulating lactate transporters (e.g., SLC16A3 knockdown or SLC16A4 upregulation) - have shown promise in alleviating this immunosuppression and enhancing immunotherapy efficacy[192-194]. Given its pivotal role in the HCC TIME, inhibiting lactate production and transport represents a promising strategy that simultaneously modulates tumor metabolism and remodels the immune landscape. Combining such approaches with immune checkpoint blockade may offer a synergistic strategy to overcome immune evasion and improve treatment responses in HCC patients[195].
Modulating the gut microbiota represents a promising frontier in liver cancer immunotherapy. As a key regulator of metabolism and immunity, the gut microbiota profoundly shapes the hepatic immune microenvironment and influences HCC development. Through the gut-liver axis, microbial metabolites - such as short-chain fatty acids (SCFAs), bile acids, and lipopolysaccharides - travel via the portal system to modulate hepatic immune homeostasis and inflammatory responses, thereby affecting TIME[196,197].
Studies indicate that HCC patients exhibit reduced gut microbial diversity, characterized by a decline in beneficial bacteria and an expansion of pro-inflammatory species. This dysbiosis contributes to local immune dysfunction and tumor immune evasion, correlating with altered immune cell infiltration, checkpoint expression, and variable responses to immunotherapy[198,199]. Interventions aimed at restoring microbial balance - including probiotics, prebiotics, fecal microbiota transplantation (FMT), or antibiotics - can reshape the TIME and enhance therapeutic efficacy[200,201]. For instance, FMT has shown potential to reverse immune tolerance and improve outcomes in patients resistant to anti-PD-1 therapy[202,203].
Furthermore, the gut microbiota regulates bile acid metabolism, which in turn influences the activity and metabolic state of hepatic immune cells and modulates immune checkpoint signaling[204,205]. Thus, targeting the gut microbiome not only helps explain heterogeneity in immunotherapy responses but also offers a novel strategy to overcome resistance. Future combinations of microbiome-based interventions with immune checkpoint blockade hold significant potential to improve survival and therapeutic outcomes in HCC.
In summary, dysregulated lactate metabolism drives immunosuppression in the HCC microenvironment, while the gut microbiota modulates the TIME through metabolic and immunologic crosstalk. Targeting lactate pathways and modulating the microbiota represent promising strategies to overcome current therapeutic limitations, enhance anti-tumor immunity, and improve outcomes in HCC, holding considerable translational potential.
Combined treatment trend: multidimensional targeted TIME
The combination of ICIs with molecular targeted agents is now a cornerstone of first-line therapy for HCC. A key regimen, atezolizumab (anti-PD-L1) plus bevacizumab (anti-VEGF), demonstrated superior efficacy over sorafenib monotherapy in the phase III IMbrave150 trial, achieving an ORR of 30% and a complete response rate of 8%. This strategy works synergistically by concurrently inhibiting angiogenesis and remodeling TIME, effectively converting immunologically “cold” tumors into “hot” ones to enhance immune cell infiltration and activity[206]. The positive correlation between VEGF pathway activity and immunosuppressive gene expression further supports the strong rationale for this combination, highlighting its significant potential to improve treatment outcomes in HCC[207].
Combining immunotherapy with local treatments such as TACE and radiotherapy represents a promising strategy for HCC[208]. TACE combined with systemic therapy has become a recommended regimen for advanced HCC, as it enhances the release of tumor-associated antigens and inflammatory cytokines, thereby activating the cancer-immunity cycle and remodeling the TIME to increase immunotherapy sensitivity[209,210]. Similarly, radiotherapy combined with ICIs can profoundly modulate the TIME, augment T cell-mediated anti-tumor immunity, and has shown encouraging efficacy in clinical settings for refractory HCC[211,212]. Other local modalities, including radiofrequency ablation, also demonstrate synergistic potential with immunotherapy in preclinical and early clinical studies. This integrated approach offers a promising avenue to improve outcomes for HCC patients[213,214].
The combination of multi-targeted TKIs with immunotherapy is an increasingly studied strategy in HCC[215]. TKIs not only directly inhibit tumor growth and angiogenesis but also remodel TIME by enhancing immune cell activity and suppressing immunosuppressive functions, thereby potentially sensitizing tumors to immunotherapy[210,216]. For instance, the phase III COSMIC-312 trial, evaluating cabozantinib plus atezolizumab in treatment-naïve advanced HCC, shows promising translational potential[210]. Similarly, the combination of lenvatinib with ICIs has demonstrated robust anti-tumor activity, prompting ongoing research into mechanisms of resistance and optimization of personalized treatment strategies[217].
Combination therapies targeting key components of the TIME, such as the gut microbiota and TAMs, are an emerging focus. For example, quercetin combined with anti-PD-1 therapy can remodel the TIME by modulating both the gut microbiome and macrophage polarization, potentially enhancing immunotherapy efficacy[218]. Furthermore, preclinical strategies co-targeting MDSCs and Tregs have shown promise in boosting anti-tumor immunity. These approaches aim to overcome immune evasion by simultaneously disrupting multiple immunosuppressive axes within the TME[219,220].
Finally, emerging technologies such as nanoparticle-based delivery systems offer innovative platforms for combinatorial treatment. By enabling targeted and synchronized drug release, nanocarriers can enhance immunogenic cell death, promote antigen release, and activate immune cells, thereby improving the precision and efficacy of immunotherapy. For example, an integrated nanoparticle platform combining ultrasound contrast with chemotherapy, photothermal, and immunotherapy has been shown in animal models to potently activate antitumor immunity and suppress metastasis[221].
Additionally, in clinical practice, the safety and tolerability of combination therapies must be carefully considered. While combining radiotherapy with other treatments has significantly improved efficacy, it also increases the risk of hematologic and liver toxicity, necessitating close monitoring and management[222]. Furthermore, factors such as sarcopenia, which is associated with treatment prognosis, indicate that combination therapy regimens should be individualized[223]. This personalized matching approach highly aligns with the etiological heterogeneity of the immune microenvironment in HCC. For instance, in HBV+ MASLD+ HCC, the presence of high CD8+ T cell infiltration and an enriched population of precursor-exhausted T cells suggests that this subgroup of patients may derive the greatest benefit from immunotherapy[113]. The mechanisms, clinical advantages, and inherent limitations of these diverse combination regimens and other emerging strategies are systematically summarized in Table 2.
Classification, mechanisms, and challenges of immunotherapy strategies targeting the HCC TIME
| Therapeutic category | Mechanism of action | Specific regimen/targets | Associated heterogeneity dimension | Key advantages | Major limitations and challenges | Ref. |
| I. Immune checkpoint inhibitors | ||||||
| Standard ICIs | Nivolumab, Pembrolizumab, Durvalumab | PD-1/PD-L1 | Temporal (Advanced stages) | Established first/second-line standard; durable response in subsets | Low ORR (15%-30%); primary/acquired resistance; irAEs | [224] |
| Ipilimumab, Tremelimumab | CTLA-4 | |||||
| Emerging ICIs | Inhibits alternative co-inhibitory receptors to prevent terminal exhaustion | LAG-3, TIGIT, VISTA, Siglec-9 | Etiology (HBV-specific exhaustion) | [H/M] Potential to overcome PD-1 resistance; synergistic with anti-PD-1 | Mostly in early clinical phases; efficacy as monotherapy is limited | [225-227] |
| II. TME remodeling strategies | ||||||
| Recruitment inhibition | Disrupts chemotactic signals to block suppressive cell infiltration | CXCR4/CXCL12 axis antagonists | Spatial (Invasive margin) | [H/M] Reduces “immune-excluded” niches; clears path for CTL infiltration | Redundancy in chemokine pathways; potential systemic side effects | [38,228] |
| Polarization modulation | Reprograms TAMs/MDSCs from suppressive to pro-inflammatory phenotypes | PTPN2 silencing, Vitamin B3 (GPR109A) | Spatial (Metabolic barrier) | [M] Directly dismantles the core immunosuppressive barrier; enhances ICD | [M] Precise liver delivery challenges; metabolic adaptability of TAMs | [229,230] |
| III. Adoptive cell therapy | ||||||
| Engineered T cells | Redirects T cells to recognize tumor-specific antigens independently of MHC | CAR-T (GPC3, AFP), BiTEs | Etiology (Antigen-specific) | High specificity; potent lysis of antigen-positive HCC cells | Antigen loss/heterogeneity; poor penetration into solid tumor core | [231,232] |
| Native/Infiltrating cells | Exploits broad antigen recognition of tumor-resident or innate killers | TILs, NK cells, T cells | Spatial (Tumor infiltration) | Better management of tumor heterogeneity than CAR-T; low GVHD risk | Labor-intensive expansion; rapid exhaustion within the inhibitory TME | [233-235] |
| Cell-derived vesicles | Leverages engineered exosomes for targeted delivery of cytotoxic cargo | CAR-Exosomes | Spatial (Deep penetration) | [M] Superior tissue penetration; lower risk of cytokine storm than live cells | [M] Standardization of isolation; short circulation half-life | [236,237] |
| IV. Metabolic and microbiome interventions | ||||||
| Metabolic regulation | Normalizes TME pH and nutrient availability to reinvigorate effector cells | LDH inhibitors, SLC16A3 knockdown | Spatial (Hypoxic core) | [M] Reverses metabolic-driven immune silencing in the tumor core | [M] Potential interference with systemic glucose metabolism | [74,238] |
| Gut-liver axis modulation | Restores microbial diversity to activate hepatic immunity via portal system | FMT, Probiotics, SCFAs | Etiology (Metabolic-related) | Systematic and non-invasive; highly relevant for MASH-related HCC | High inter-individual variability; difficult to standardize across cohorts | [239,240] |
| V. Multidimensional combination strategies | ||||||
| ICI + Anti-angiogenic | Normalizes vasculature to increase oxygenation and CTL infiltration | Atezolizumab + Bevacizumab | Spatial (Vascular barrier) | Current gold standard; simultaneously addresses hypoxia and exclusion | Risk of bleeding/hemorrhage; vascular pruning may lead to secondary hypoxia | [241,242] |
| ICI + Local-treatment | Triggers ICD to release “in situ” vaccines | TACE, Radiotherapy, RFA | Temporal (Window of activation) | Creates a temporal window for priming; converts “cold” tumors to “hot” | Optimal sequencing remains unclear; potential treatment-induced toxicity | [243] |
CONCLUSION
This review highlights that the inhibitory TIME of HCC acts as one of the key drivers of disease progression and a major obstacle to effective immunotherapy. Actively shaped by a network of suppressive cells, including TAMs, MDSCs, and Tregs - the TIME impairs effector lymphocyte function and promotes immune evasion through diverse molecular and metabolic mechanisms. Crucially, the substantial heterogeneity of this immunosuppression, which manifests across divergent etiological backgrounds, spatial topologies, and temporal evolutionary trajectories, partly underlies the highly variable patient responses to treatments such as ICIs.
While therapeutic strategies increasingly aim to target and remodel the TIME - with combination therapies demonstrating notable clinical success - resistance, which is partly rooted in the adaptability of this immunosuppressive network, remains a major clinical challenge. Currently, research on the HCC TIME is transitioning from macroscopic, static descriptions of cellular populations to microscopic, dynamic analyses of spatial interactions. However, critical gaps in knowledge persist. These include that the distinct immune heterogeneity driven by different etiologies remains incompletely understood; precise characterization mapping the spatial topology and dynamic evolutionary trajectories of the TIME requires further exploration; and the metabolic-immune interactive network, particularly the metabolic competition between immune cells within the hypoxic microenvironment, remains incompletely deciphered.
Addressing these gaps and unlocking the full therapeutic potential of immunotherapy would benefit from a paradigm shift in future research. Efforts should focus on leveraging cutting-edge spatial multi-omics technologies to construct high-resolution, three-dimensional maps of the TIME. Furthermore, identifying robust predictive biomarkers is urgently needed to help precisely guide clinical interventions. Future translational research should also systematically investigate the mechanisms of microenvironmental remodeling designed to reverse “cold tumors” through rational combined local-regional therapies and immunomodulators. Simultaneously, exploring strategies that target innate immunity, investigate novel immune checkpoints, and develop microbiome-based interventions via the gut-liver axis will be important. Ultimately, deciphering the dynamic regulation of the TIME may provide a robust theoretical foundation for developing next-generation combinatorial approaches, sustainably reinvigorating anti-tumor immunity, and facilitating the realization of personalized precision medicine in HCC.
DECLARATIONS
Acknowledgments
The Graphical Abstract was created with BioRender.com.
Authors’ contributions
Developed the concept and designed the review: Mao T, Jiang J, Liu P
Independently screened the full texts of eligible studies to confirm compliance with inclusion criteria, assess study quality, and extract data: Mao T, Jiang J, Liu P, Zhao Y, Zhong L, Zhang Z, Wu K, Xia L
Provided overall supervision and revision of the manuscript: Xia L
Contributed to manuscript drafting and revision, and approved the final version for submission: Mao T, Jiang J, Liu P, Zhao Y, Zhong L, Zhang Z, Wu K, Xia L
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This research was supported by grants from the National Natural Science Foundation of China (Nos. 82525045 and U23A20451, Xia L) and the Basic Research Support Program of Huazhong University of Science and Technology (No. 2023BR038, Xia L).
Conflicts of interest
Wu K and Xia L are the Associate Chief Editors of the journal Hepatoma Research. They were not involved in any stage of the editorial process, including reviewer selection, manuscript handling, or decision-making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
2. Han B, Zheng R, Zeng H, et al. Cancer incidence and mortality in China, 2022. J Natl Cancer Cent. 2024;4:47-53.
3. Rimassa L, Finn RS, Sangro B. Combination immunotherapy for hepatocellular carcinoma. J Hepatol. 2023;79:506-15.
4. Zheng J, Shao M, Yang W, Ren J, Chen X, Yang H. Benefits of combination therapy with immune checkpoint inhibitors and predictive role of tumour mutation burden in hepatocellular carcinoma: a systematic review and meta-analysis. Int Immunopharmacol. 2022;112:109244.
5. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19:151-72.
6. Zheng J, Wang S, Xia L, et al. Hepatocellular carcinoma: signaling pathways and therapeutic advances. Signal Transduct Target Ther. 2025;10:35.
7. Liu Y, Xun Z, Ma K, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78:770-82.
8. Wang H, Liang Y, Liu Z, et al. POSTN+ cancer-associated fibroblasts determine the efficacy of immunotherapy in hepatocellular carcinoma. J Immunother Cancer. 2024;12:e008721.
9. Yang C, Geng H, Yang X, et al. Targeting the immune privilege of tumor-initiating cells to enhance cancer immunotherapy. Cancer Cell. 2024;42:2064-81.e19.
10. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221:107753.
11. Bejarano L, Jordāo MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021;11:933-59.
12. Gottwick C, Carambia A, Herkel J. Harnessing the liver to induce antigen-specific immune tolerance. Semin Immunopathol. 2022;44:475-84.
13. Henriques-Pons A, Vacani-Martins N, Dos Santos CLP, Meuser-Batista M. The liver’s dilemma: sensing real danger in a sea of PAMPs: the (arterial) sinusoidal segment theory. Front Immunol. 2024;15:1503063.
14. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. 2010;10:753-66.
17. Balogh J, Victor D 3rd, Asham EH, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma. 2016;3:41-53.
18. Baumert TF, Jühling F, Ono A, Hoshida Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med. 2017;15:52.
19. Kudo M. Changing the treatment paradigm for hepatocellular carcinoma using atezolizumab plus bevacizumab combination therapy. Cancers. 2021;13:5475.
20. Jiang X, Wang J, Deng X, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10.
21. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49-61.
22. Wang D, Yang L, Yue D, et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019;452:244-53.
23. Yang CL, Song R, Hu JW, et al. Integrating single-cell and bulk RNA sequencing reveals CK19 + cancer stem cells and their specific SPP1 + tumor-associated macrophage niche in HBV-related hepatocellular carcinoma. Hepatol Int. 2024;18:73-90.
24. Chen S, Zhang P, Zhu G, et al. Targeting GSDME-mediated macrophage polarization for enhanced antitumor immunity in hepatocellular carcinoma. Cell Mol Immunol. 2024;21:1505-21.
25. Li L, Sun P, Zhang C, Li Z, Cui K, Zhou W. MiR-98 modulates macrophage polarization and suppresses the effects of tumor-associated macrophages on promoting invasion and epithelial-mesenchymal transition of hepatocellular carcinoma. Cancer Cell Int. 2018;18:95.
26. Lu Y, Sun Q, Guan Q, et al. The XOR-IDH3α axis controls macrophage polarization in hepatocellular carcinoma. J Hepatol. 2023;79:1172-84.
27. Jiang Y, Han Q, Zhao H, Zhang J. Promotion of epithelial-mesenchymal transformation by hepatocellular carcinoma-educated macrophages through Wnt2b/β-catenin/c-Myc signaling and reprogramming glycolysis. J Exp Clin Cancer Res. 2021;40:13.
28. Gao Z, Li XG, Feng SR, et al. Autophagy suppression facilitates macrophage M2 polarization via increased instability of NF-κB pathway in hepatocellular carcinoma. Int Immunopharmacol. 2023;123:110685.
29. Lu CS, Shiau AL, Su BH, et al. Oct4 promotes M2 macrophage polarization through upregulation of macrophage colony-stimulating factor in lung cancer. J Hematol Oncol. 2020;13:62.
30. Wei CY, Zhu MX, Zhang PF, et al. PKCα/ZFP64/CSF1 axis resets the tumor microenvironment and fuels anti-PD1 resistance in hepatocellular carcinoma. J Hepatol. 2022;77:163-76.
31. Feng R, Cui Z, Yang L, Liu Z. Sphingosine 1-phosphate derived from tumor-educated hepatic stellate cells combining with S1PR4 promotes tumor associated macrophages differentiation through FAO modulation. Sci Rep. 2025;15:20507.
32. Kim DH, Kang YN, Jin J, et al. Glutamine-derived aspartate is required for eIF5A hypusination-mediated translation of HIF-1α to induce the polarization of tumor-associated macrophages. Exp Mol Med. 2024;56:1123-36.
33. Zhang X, Lao M, Sun K, et al. Sphingolipid synthesis in tumor-associated macrophages confers immunotherapy resistance in hepatocellular carcinoma. Sci Adv. 2025;11:eadv0558.
34. Yang X, Deng B, Zhao W, et al. FABP5+ lipid-loaded macrophages process tumour-derived unsaturated fatty acid signal to suppress T-cell antitumour immunity. J Hepatol. 2025;82:676-89.
35. Cai J, Zhang P, Cai Y, et al. Lactylation-driven NUPR1 promotes immunosuppression of tumor-infiltrating macrophages in hepatocellular carcinoma. Adv Sci. 2025;12:e2413095.
36. Yu H, Pan J, Zheng S, et al. Hepatocellular carcinoma cell-derived exosomal miR-21-5p induces macrophage M2 polarization by targeting RhoB. Int J Mol Sci. 2023;24:4593.
37. Li X, Yao W, Yuan Y, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157-67.
38. Chen J, Feng W, Sun M, et al. TGF-β1-induced SOX18 elevation promotes hepatocellular carcinoma progression and metastasis through transcriptionally upregulating PD-L1 and CXCL12. Gastroenterology. 2024;167:264-80.
39. Piqué-Gili M, Andreu-Oller C, Mesropian A, et al. Oncogenic role of PMEPA1 and its association with immune exhaustion and TGF-β activation in HCC. JHEP Rep. 2024;6:101212.
40. Xie M, Lin Z, Ji X, et al. FGF19/FGFR4-mediated elevation of ETV4 facilitates hepatocellular carcinoma metastasis by upregulating PD-L1 and CCL2. J Hepatol. 2023;79:109-25.
41. Wang D, Li X, Li J, et al. APOBEC3B interaction with PRC2 modulates microenvironment to promote HCC progression. Gut. 2019;68:1846-57.
42. Nosaka T, Ohtani M, Yamashita J, et al. PD-L1+ tumor-associated macrophages induce CD8+ T Cell exhaustion in hepatocellular carcinoma. Neoplasia. 2025;69:101234.
43. Liu Z, Wang Y, Dou C, et al. Hypoxia-induced up-regulation of VASP promotes invasiveness and metastasis of hepatocellular carcinoma. Theranostics. 2018;8:4649-63.
44. Wei X, Wang H, Liu H, et al. Disruption of tumor-intrinsic PGAM5 increases anti-PD-1 efficacy through the CCL2 signaling pathway. J Immunother Cancer. 2025;13:e009993.
45. Zhang W, Liu Y, Yan Z, et al. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J Immunother Cancer. 2020;8:e000285.
46. Sun Y, Zhou P, Qian J, et al. Spermine synthase engages in macrophages M2 polarization to sabotage antitumor immunity in hepatocellular carcinoma. Cell Death Differ. 2025;32:573-86.
47. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21:485-98.
48. Ceredig R, Rolink AG, Brown G. Models of haematopoiesis: seeing the wood for the trees. Nat Rev Immunol. 2009;9:293-300.
49. Goldmann O, Medina E. Metabolic pathways fueling the suppressive activity of myeloid-derived suppressor cells. Front Immunol. 2024;15:1461455.
50. Alicea-Torres K, Sanseviero E, Gui J, et al. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat Commun. 2021;12:1717.
51. Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150.
53. Törnell A, Blick E, Al-Dury S, et al. Presence of MDSC associates with impaired antigen-specific T cell reactivity following COVID-19 vaccination in cirrhotic patients. Front Immunol. 2023;14:1287287.
54. Chivite-Lacaba M, Justo I, Utrero-Rico A, et al. Delineation of monocytic and early-stage myeloid-derived suppressor cells in the peripheral blood of patients with hepatocarcinoma. Int J Cancer. 2025;156:2416-28.
55. Santagata S, Rea G, Castaldo D, et al. Hepatocellular carcinoma (HCC) tumor microenvironment is more suppressive than colorectal cancer liver metastasis (CRLM) tumor microenvironment. Hepatol Int. 2024;18:568-81.
56. Wu X, Pan B, Chu C, et al. CXCL16/CXCR6/TGF-β feedback loop between M-MDSCs and Treg inhibits anti-bacterial immunity during biofilm infection. Adv Sci. 2025;12:e2409537.
57. Noman MZ, Desantis G, Janji B, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781-90.
58. Zhang Z, Huang W, Hu D, et al. E-twenty-six-specific sequence variant 5 (ETV5) facilitates hepatocellular carcinoma progression and metastasis through enhancing polymorphonuclear myeloid-derived suppressor cell (PMN-MDSC)-mediated immunosuppression. Gut. 2025;74:1137-49.
59. Xia S, Wu J, Zhou W, et al. SLC7A2 deficiency promotes hepatocellular carcinoma progression by enhancing recruitment of myeloid-derived suppressors cells. Cell Death Dis. 2021;12:570.
60. Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T cells. Gastroenterology. 2008;135:234-43.
61. Yashaswini CN, Qin T, Bhattacharya D, et al. Phenotypes and ontogeny of senescent hepatic stellate cells in metabolic dysfunction-associated steatohepatitis. J Hepatol. 2024;81:207-17.
62. Gavin MA, Rasmussen JP, Fontenot JD, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771-5.
63. Tan SN, Hao J, Ge J, et al. Regulatory T cells converted from Th1 cells in tumors suppress cancer immunity via CD39. J Exp Med. 2025;222:e20240445.
64. Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, Li Z. Transforming growth factor-β1 in regulatory T cell biology. Sci Immunol. 2022;7:eabi4613.
65. Shiri AM, Fard-Aghaie M, Bedke T, et al. Foxp3 + Treg-derived IL-10 promotes colorectal cancer-derived lung metastasis. Sci Rep. 2024;14:30483.
66. Sawant DV, Yano H, Chikina M, et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. 2019;20:724-35.
67. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U S A. 2021;118:e2023739118.
68. Huang CT, Workman CJ, Flies D, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503-13.
69. Pan B, Yao Y, Wu H, et al. N-glycosylated LTβR increases the Th17/Treg cell ratio in liver cancer by blocking RORC ubiquitination and FOXP3 transcription. Cell Death Dis. 2025;16:421.
70. Li Q, Zhang L, You W, et al. PRDM1/BLIMP1 induces cancer immune evasion by modulating the USP22-SPI1-PD-L1 axis in hepatocellular carcinoma cells. Nat Commun. 2022;13:7677.
71. Gao Y, You M, Fu J, et al. Intratumoral stem-like CCR4+ regulatory T cells orchestrate the immunosuppressive microenvironment in HCC associated with hepatitis B. J Hepatol. 2022;76:148-59.
72. Li S, Xia H, Wang Z, et al. Intratumoral microbial heterogeneity affected tumor immune microenvironment and determined clinical outcome of HBV-related HCC. Hepatology. 2023;78:1079-91.
73. Ding J, Liu H, Zhang X, et al. Integrative multiomic analysis identifies distinct molecular subtypes of NAFLD in a Chinese population. Sci Transl Med. 2024;16:eadh9940.
74. Nong Y, Chen X, Zhang B, et al. M2 macrophages and tumor cells engage in a metabolic feedback loop to drive HCC progression. Sci Rep. 2025;15:41701.
75. Qin SK, Li Q, Ming Xu J, et al. Icaritin-induced immunomodulatory efficacy in advanced hepatitis B virus-related hepatocellular carcinoma: immunodynamic biomarkers and overall survival. Cancer Sci. 2020;111:4218-31.
76. Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP+CTLA-4+Foxp3+ T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013;73:2435-44.
77. Li A, Ji B, Yang Y, et al. Single-cell RNA sequencing highlights the role of PVR/PVRL2 in the immunosuppressive tumour microenvironment in hepatocellular carcinoma. Front Immunol. 2023;14:1164448.
78. Wen J, Zhang X, Wong CC, et al. Targeting squalene epoxidase restores anti-PD-1 efficacy in metabolic dysfunction-associated steatohepatitis-induced hepatocellular carcinoma. Gut. 2024;73:2023-36.
79. Gu X, Wei H, Suo C, et al. Itaconate promotes hepatocellular carcinoma progression by epigenetic induction of CD8+ T-cell exhaustion. Nat Commun. 2023;14:8154.
80. Huang J, Tsang WY, Fang XN, et al. FASN inhibition decreases MHC-I degradation and synergizes with PD-L1 checkpoint blockade in hepatocellular carcinoma. Cancer Res. 2024;84:855-71.
81. Barends M, Koller N, Schölz C, et al. Dynamic interactome of the MHC I peptide loading complex in human dendritic cells. Proc Natl Acad Sci U S A. 2023;120:e2219790120.
82. Zhang CY, Liu S, Yang M. Nutrition deprivation affects the cytotoxic effect of CD8 T cells in hepatocellular carcinoma. World J Gastrointest Oncol. 2022;14:1887-91.
83. Bai X, Zhou Y, Yokota Y, et al. Adaptive antitumor immune response stimulated by bio-nanoparticle based vaccine and checkpoint blockade. J Exp Clin Cancer Res. 2022;41:132.
84. Eberhardt CS, Kissick HT, Patel MR, et al. Functional HPV-specific PD-1+ stem-like CD8 T cells in head and neck cancer. Nature. 2021;597:279-84.
85. Huang H, Pan Y, Mai Q, et al. Targeting CDCP1 boost CD8+ T cells-mediated cytotoxicity in cervical cancer via the JAK/STAT signaling pathway. J Immunother Cancer. 2024;12:e009416.
86. Zhao L, Yuan H, Wang Y, et al. p-STAT3-elevated E3 ubiquitin ligase DTX4 confers the stability of HBV cccDNA by ubiquitinating APOBEC3B in liver. Theranostics. 2024;14:6036-52.
87. De Simone V, Franzè E, Ronchetti G, et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene. 2015;34:3493-503.
88. Wang T, Fan Y, Tan S, et al. Probiotics and their metabolite spermidine enhance IFN-γ+CD4+ T cell immunity to inhibit hepatitis B virus. Cell Rep Med. 2024;5:101822.
89. Mohammadi-Kordkhayli M, Sahraian MA, Ghorbani S, et al. Vitamins A and D enhance the expression of Ror-γ-targeting miRNAs in a mouse model of multiple sclerosis. Mol Neurobiol. 2023;60:5853-65.
90. Gharibi T, Barpour N, Hosseini A, et al. STA-21, a small molecule STAT3 inhibitor, ameliorates experimental autoimmune encephalomyelitis by altering Th-17/Treg balance. Int Immunopharmacol. 2023;119:110160.
91. Motavalli R, Soltani-Zangbar MS, Fereydoonzadeh K, et al. Evaluation of T helper17 as skeletal homeostasis factor in peripheral blood mononuclear cells and T helper cells of end-stage renal disease cases with impaired parathyroid hormone. Mol Biol Rep. 2023;50:4097-104.
92. Wang H, Herman A, Barrow F, et al. Single-cell RNA sequencing reveals a reprogramming of hepatic immune cells and a protective role for B cells in MASH-driven HCC. Hepatol Commun. 2025;9:e0668.
93. Cinnamon E, Stein I, Zino E, et al. RORc-expressing immune cells negatively regulate tertiary lymphoid structure formation and support their pro-tumorigenic functions. J Hepatol. 2025;82:1050-67.
94. Morsing AE, Green K, Clausen S, et al. Age-associated B cells enhance tertiary lymphoid structures in the liver and promote HCC formation. Hepatol Commun. 2025;9:e0816.
95. Lian SL, Lu YT, Lu YJ, Yao YL, Wang XL, Jiang RQ. Tumor-associated macrophages promoting PD-L1 expression in infiltrating B cells through the CXCL12/CXCR4 axis in human hepatocellular carcinoma. Am J Cancer Res. 2024;14:832-53.
96. Zhang B, Vogelzang A, Miyajima M, et al. B cell-derived GABA elicits IL-10+ macrophages to limit anti-tumour immunity. Nature. 2021;599:471-6.
97. Chen Z, Zhang G, Ren X, et al. Cross-talk between myeloid and B cells shapes the distinct microenvironments of primary and secondary liver cancer. Cancer Res. 2023;83:3544-61.
98. Zou J, Luo C, Xin H, et al. The role of tumor-infiltrating B cells in the tumor microenvironment of hepatocellular carcinoma and its prognostic value: a bioinformatics analysis. J Gastrointest Oncol. 2022;13:1959-66.
99. Schaafsma E, Jiang C, Cheng C. B cell infiltration is highly associated with prognosis and an immune-infiltrated tumor microenvironment in neuroblastoma. J Cancer Metastasis Treat. 2021;7:34.
100. Tavakoli S, Samareh-Salavati M, Abdolahi S, et al. Cell therapy using anti-NKG2A pretreated natural killer cells in patients with hepatocellular carcinoma. Adv Pharm Bull. 2024;14:918-26.
101. Yu L, Liu X, Wang X, et al. TIGIT+ TIM-3+ NK cells are correlated with NK cell exhaustion and disease progression in patients with hepatitis B virus‑related hepatocellular carcinoma. Oncoimmunology. 2021;10:1942673.
102. Xi F, Sun H, Peng H, et al. Hepatocyte-derived FGL1 accelerates liver metastasis and tumor growth by inhibiting CD8+ T and NK cells. JCI Insight. 2024;9:e173215.
103. Cheng X, Tan X, Wang W, et al. Long-Chain acylcarnitines induce senescence of invariant natural killer T cells in hepatocellular carcinoma. Cancer Res. 2023;83:582-94.
104. Han J, Ke C, Jiang B, Zhou H, Xu H, Xie X. Down-regulation of PR/SET domain 10 underlies natural killer cell dysfunction in hepatocellular carcinoma. Clin Exp Immunol. 2021;206:366-77.
105. Cui C, Fu K, Yang L, et al. Hypoxia-inducible gene 2 promotes the immune escape of hepatocellular carcinoma from nature killer cells through the interleukin-10-STAT3 signaling pathway. J Exp Clin Cancer Res. 2019;38:229.
106. Shin SK, Oh S, Chun SK, et al. Immune signature and therapeutic approach of natural killer cell in chronic liver disease and hepatocellular carcinoma. J Gastroenterol Hepatol. 2024;39:1717-27.
107. MacNabb BW, Chen X, Tumuluru S, et al. Dendritic cells can prime anti-tumor CD8+ T cell responses through major histocompatibility complex cross-dressing. Immunity. 2022;55:2206-8.
108. Li T, Li B, Lin L, et al. Anti-CTLA-4 antibody self-presented dendritic cell nanovesicles boost the immunotherapy of hepatocellular carcinoma after microwave ablation. J Control Release. 2024;376:913-29.
109. Yao F, Yuan Q, Yan Y, et al. Yu-Ping-Feng-San improve the immunosuppression of microenvironment in hepatocellular carcinoma by promoting the maturation of DCs through the JAK2-STAT3 pathway. Sci Rep. 2024;14:31522.
110. Li J, Lin W, Huang T, Chen M, Lin Q. IL-12 improves the anti-HCC efficacy of dendritic cells loaded with exosomes from overexpressing Rab27a tumor cells. Exp Cell Res. 2024;439:114073.
111. Wang C, Huang X, Wu Y, Wang J, Li F, Guo G. Tumor cell-associated exosomes robustly elicit anti-tumor immune responses through modulating dendritic cell vaccines in lung tumor. Int J Biol Sci. 2020;16:633-43.
112. Zhang S, Xu H, Li M, et al. An etiology-stratified single-cell atlas identifies FABP4 as a prognostic marker for MASLD-related HCC. J Hepatol. 2026;84:920-32.
113. He J, Liu B, Zhao W, et al. The glucose sensor NSUN2-m5C modification regulates tumor-immune glucose metabolism reprogramming to drive hepatocellular carcinoma evolution. Int J Biol Sci. 2025;21:4529-48.
114. Sun Y, Wu P, Zhang Z, et al. Integrated multi-omics profiling to dissect the spatiotemporal evolution of metastatic hepatocellular carcinoma. Cancer Cell. 2024;42:135-56.e17.
115. Jang EJ, Choi HJ, You YK, et al. Differential infiltration of T-cell populations in tumor and liver tissues predicts recurrence-free survival in surgically resected hepatocellular carcinoma. Cancers. 2025;17:1548.
116. Wu WJ, Wang J, Chen F, et al. Exploration of heterogeneity and recurrence signatures in hepatocellular carcinoma. Mol Oncol. 2025;19:2388-411.
117. Li J, Xu H, Han J, et al. Lymphocyte function in tertiary lymphoid structures predicts hepatocellular carcinoma outcome. Lab Invest. 2024;104:102144.
118. Rennert C, Tauber C, Fehrenbach P, et al. Adaptive subsets limit the anti-tumoral NK-cell activity in hepatocellular carcinoma. Cells. 2021;10:1369.
119. Liu H, Zhao R, Qin R, et al. Panoramic comparison between NK cells in healthy and cancerous liver through single-cell RNA sequencing. Cancer Biol Med. 2022;19:1334-51.
120. Classon P, Wixom AQ, Calixto Mancipe N, et al. Role of long-chain acyl-CoA synthetases in MASH-driven hepatocellular carcinoma and ferroptosis. Am J Physiol Gastrointest Liver Physiol. 2025;329:G571-84.
121. Li S, Zhang J, Wei W, Zhang Z, Huang W, Xia L. The important role of myeloid-derived suppressor cells: from hepatitis to liver cancer. Biochim Biophys Acta Rev Cancer. 2025;1880:189329.
122. Wang H, Zhang H, Wang Y, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. 2021;75:1271-83.
123. Tan J, Fan W, Liu T, et al. TREM2+ macrophages suppress CD8+ T-cell infiltration after transarterial chemoembolisation in hepatocellular carcinoma. J Hepatol. 2023;79:126-40.
124. Shao Y, Lo CM, Ling CC, et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett. 2014;355:264-72.
125. Kotsiliti E, Leone V, Schuehle S, et al. Intestinal B cells license metabolic T-cell activation in NASH microbiota/antigen-independently and contribute to fibrosis by IgA-FcR signalling. J Hepatol. 2023;79:296-313.
126. Lin C, Chen Y, Zhang F, Zhu P, Yu L, Chen W. Single-cell RNA sequencing reveals the mediatory role of cancer-associated fibroblast PTN in hepatitis B virus cirrhosis-HCC progression. Gut Pathog. 2023;15:26.
127. Lu Y, Liu Y, Zuo X, et al. CXCL12+ tumor-associated endothelial cells promote immune resistance in hepatocellular carcinoma. J Hepatol. 2025;82:634-48.
128. Li W, Chen G, Peng H, et al. Research progress on dendritic cells in hepatocellular carcinoma immune microenvironments. Biomolecules. 2024;14:1161.
129. Han Y, Chen Z, Yang Y, et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. 2014;59:567-79.
130. Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253-7.
131. Li L, Wang H. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 2016;379:191-7.
132. Ji Z, Li J, Zhang S, Jia Y, Zhang J, Guo Z. The load of hepatitis B virus reduces the immune checkpoint inhibitors efficiency in hepatocellular carcinoma patients. Front Immunol. 2024;15:1480520.
133. Gallage S, Ali A, Barragan Avila JE, et al. A 5:2 intermittent fasting regimen ameliorates NASH and fibrosis and blunts HCC development via hepatic PPARα and PCK1. Cell Metab. 2024;36:1371-93.e7.
134. Lu J, Chan TT, Wang Y, et al. FADD activation in hepatocellular carcinoma potentiates CD8+ T-cell responses and sensitizes to immune checkpoint inhibitors. Cancer Res. 2025;85:3454-70.
135. Gao Y, Gong Y, Song X, et al. Dihydroartemisinin inhibits histone lactylation through YAP1 to act as a ‘hot’ switch for ‘cold’ tumor in hepatocellular carcinoma. Phytomedicine. 2025;148:157307.
136. Xiao R, Tian Y, Zhang J, et al. Increased Siglec-9/Siglec-9L interactions on NK cells predict poor HCC prognosis and present a targetable checkpoint for immunotherapy. J Hepatol. 2024;80:792-804.
137. Jiang Z, Wu Y, Miao Y, et al. HCCDB v2.0: decompose expression variations by single-cell RNA-seq and spatial transcriptomics in HCC. Genomics Proteomics Bioinformatics. 2024;22:qzae011.
138. Dong S, Guo X, Han F, He Z, Wang Y. Emerging role of natural products in cancer immunotherapy. Acta Pharm Sin B. 2022;12:1163-85.
139. Chen ZQ, Zuo XL, Cai J, et al. Hypoxia-associated circPRDM4 promotes immune escape via HIF-1α regulation of PD-L1 in hepatocellular carcinoma. Exp Hematol Oncol. 2023;12:17.
140. Long S, Li M, Chen J, et al. Spatial patterns and MRI-based radiomic prediction of high peritumoral tertiary lymphoid structure density in hepatocellular carcinoma: a multicenter study. J Immunother Cancer. 2024;12:e009879.
141. Li H, Liu H, Fu H, et al. Peritumoral tertiary lymphoid structures correlate with protective immunity and improved prognosis in patients with hepatocellular carcinoma. Front Immunol. 2021;12:648812.
142. Prawira A, Xu H, Mei Y, et al. Targeting Treg-fibroblast interaction to enhance immunotherapy in steatotic liver disease-related hepatocellular carcinoma. Gut. 2025;75:105-18.
143. Wang L, Zhu L, Liang C, et al. Targeting N6-methyladenosine reader YTHDF1 with siRNA boosts antitumor immunity in NASH-HCC by inhibiting EZH2-IL-6 axis. J Hepatol. 2023;79:1185-200.
144. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19:222-32.
145. Li X, Ramadori P, Pfister D, Seehawer M, Zender L, Heikenwalder M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat Rev Cancer. 2021;21:541-57.
146. Sun Y, Wu L, Zhong Y, et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell. 2021;184:404-21.e16.
147. Calderaro J, Petitprez F, Becht E, et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol. 2019;70:58-65.
148. Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450-6.
149. Chen DP, Ning WR, Jiang ZZ, et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J Hepatol. 2019;71:333-43.
150. Cappuyns S, Piqué-Gili M, Esteban-Fabró R, et al. Single-cell RNA sequencing-derived signatures define response patterns to atezolizumab + bevacizumab in advanced hepatocellular carcinoma. J Hepatol. 2025;82:1036-49.
151. Fulgenzi CAM, Cheon J, D’Alessio A, et al. Reproducible safety and efficacy of atezolizumab plus bevacizumab for HCC in clinical practice: results of the AB-real study. Eur J Cancer. 2022;175:204-13.
152. Gordan JD, Kennedy EB, Abou-Alfa GK, et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline update. J Clin Oncol. 2024;42:1830-50.
153. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology. 2017;6:e1261779.
154. Johnston RJ, Comps-Agrar L, Hackney J, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. 2014;26:923-37.
155. Kim TK, Han X, Hu Q, et al. PD-1H/VISTA mediates immune evasion in acute myeloid leukemia. J Clin Invest. 2024;134:e164325.
156. Schaafsma E, Croteau W, ElTanbouly M, et al. VISTA targeting of T-cell quiescence and myeloid suppression overcomes adaptive resistance. Cancer Immunol Res. 2023;11:38-55.
157. Zhang J, Yin Y, Tang J, Zhang Y, Tian Y, Sun F. Changes in serum interleukin-8 levels predict response to immune checkpoint inhibitors immunotherapy in unresectable hepatocellular carcinoma patients. J Inflamm Res. 2024;17:3397-406.
158. Hong C, Dong HZ, Li RN, et al. Predictive value of the hepatic immune predictive index for patients with primary liver cancer treated with immune checkpoint inhibitors. Dig Dis. 2023;41:422-30.
159. Hung HC, Lee JC, Wang YC, et al. Response prediction in immune checkpoint inhibitor immunotherapy for advanced hepatocellular carcinoma. Cancers. 2021;13:1607.
160. Yuen VW, Chiu DK, Law CT, et al. Using mouse liver cancer models based on somatic genome editing to predict immune checkpoint inhibitor responses. J Hepatol. 2023;78:376-89.
161. Budhu A, Pehrsson EC, He A, et al. Tumor biology and immune infiltration define primary liver cancer subsets linked to overall survival after immunotherapy. Cell Rep Med. 2023;4:101052.
162. Ren Z, Wang Y, Jiang D, et al. PD1+ Treg cell remodeling promotes immune homeostasis within peripheral blood and tumor microenvironment after microparticles-transarterial chemoembolization in hepatocellular carcinoma. Cancer Immunol Immunother. 2025;74:109.
163. Yang P, Qin H, Li Y, et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat Commun. 2022;13:5782.
164. Liu QQ, Li HZ, Li SX, et al. CD36-mediated accumulation of MDSCs exerts abscopal immunosuppressive responses in hepatocellular carcinoma after insufficient microwave ablation. Biochim Biophys Acta Mol Basis Dis. 2024;1870:167493.
165. Wang F, You H, Liu H, et al. Silencing PTPN2 with nanoparticle-delivered small interfering RNA remodels tumor microenvironment to sensitize immunotherapy in hepatocellular carcinoma. Acta Pharm Sin B. 2025;15:2915-29.
166. Liu Y, Yang S, Zhou Q, et al. Nanobubble-based anti-hepatocellular carcinoma therapy combining immune check inhibitors and sonodynamic therapy. Nanoscale Adv. 2022;4:4847-62.
167. Yang Y, Pei T, Hu X, et al. Dietary vitamin B3 supplementation induces the antitumor immunity against liver cancer via biased GPR109A signaling in myeloid cell. Cell Rep Med. 2024;5:101718.
168. Liu Q, Liang Z, Wang J, et al. Mannose-modified multifunctional iron-based nanozyme for hepatocellular carcinoma treatment by remodeling the tumor microenvironment. Colloids Surf B Biointerfaces. 2025;250:114548.
169. Yan Y, Hu J, Han N, et al. Sorafenib-loaded metal-organic framework nanoparticles for anti-hepatocellular carcinoma effects through synergistically potentiating ferroptosis and remodeling tumor immune microenvironment. Mater Today Bio. 2025;32:101848.
170. Kong M, Qiu L. Coordinated modulation of glucose metabolism and immunity via metal-drug nanovesicles for hepatocellular carcinoma therapy. J Control Release. 2025;384:113957.
171. Hu H, Ning S, Liu F, et al. Hafnium metal-organic framework-based glutamine metabolism disruptor for potentiating radio-immunotherapy in MYC-amplified hepatocellular carcinoma. ACS Appl Mater Interfaces. 2025;17:19367-81.
172. Nie Y. Breaking barriers: the emerging promise of CAR-T cell therapy in solid tumors. Holist Integ Oncol. 2025;4:205.
173. Liu H, Xu Y, Xiang J, et al. Targeting alpha-fetoprotein (AFP)-MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res. 2017;23:478-88.
174. Sun L, Gao F, Gao Z, et al. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J Immunother Cancer. 2021;9:e001875.
175. Steffin D, Ghatwai N, Montalbano A, et al. Interleukin-15-armoured GPC3 CAR T cells for patients with solid cancers. Nature. 2025;637:940-6.
176. Shi D, Shi Y, Kaseb AO, et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clin Cancer Res. 2020;26:3979-89.
177. Yang Z, Cheng C, Li Z, et al. Advancing liver cancer treatment with dual-targeting CAR-T therapy. J Nanobiotechnology. 2025;23:462.
178. Niu X, Zhang P, Dai L, et al. Flagellin engineering enhances CAR-T cell function by reshaping tumor microenvironment in solid tumors. J Immunother Cancer. 2025;13:e010237.
179. Nishimoto KP, Lamture G, Chanthery Y, et al. ADI-270: an armored allogeneic gamma delta T cell therapy designed to target CD70-expressing solid and hematologic malignancies. J Immunother Cancer. 2025;13:e011704.
180. Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907-12.
181. Mullinax JE, Hall M, Prabhakaran S, et al. Combination of ipilimumab and adoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic melanoma. Front Oncol. 2018;8:44.
182. Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550-7.
183. Park H, Kim G, Kim N, Ha S, Yim H. Efficacy and safety of natural killer cell therapy in patients with solid tumors: a systematic review and meta-analysis. Front Immunol. 2024;15:1454427.
184. Fujisaki H, Kakuda H, Shimasaki N, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69:4010-7.
185. Zhang Y, Wallace DL, de Lara CM, et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology. 2007;121:258-65.
186. Zhao X, Zhao B, Sun Y, Liu A. CAR-exosomes derived from immune cells: an emerging nanoscale vanguard in overcoming tumor immunotherapy hurdles. Front Immunol. 2025;16:1655095.
187. Tong T, Gao W, Jian H, et al. The role and potential mechanisms of exosomes in the progression of hepatocellular carcinoma. Holist Integ Oncol. 2025;4:171.
189. Quinn WJ 3rd, Jiao J, TeSlaa T, et al. Lactate limits T cell proliferation via the NAD(H) redox state. Cell Rep. 2020;33:108500.
190. Bohn T, Rapp S, Luther N, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. 2018;19:1319-29.
191. Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63.
192. Jiang L, Shen Y, Feng Z, et al. The multidimensional role of SLC16A4 in hepatocellular carcinoma in silico analysis: prognostic significance, metabolic pathways, and immune microenvironment regulation. Eur J Med Res. 2025;30:1007.
193. Shen J, Wu Z, Zhou Y, et al. Knockdown of SLC16A3 decreases extracellular lactate concentration in hepatocellular carcinoma, alleviates hypoxia and induces ferroptosis. Biochem Biophys Res Commun. 2024;733:150709.
194. Li J, Zhang Y, Deng J, et al. Cross-talk between lactate metabolism and immunity reveals CEP55 as a potential regulator in the immunosuppressive microenvironment of hepatocellular carcinoma. Genes Dis. 2025;12:101399.
195. Ding CH, Yan FZ, Xu BN, et al. PRMT3 drives PD-L1-mediated immune escape through activating PDHK1-regulated glycolysis in hepatocellular carcinoma. Cell Death Dis. 2025;16:158.
196. Che Y, Chen G, Guo Q, Duan Y, Feng H, Xia Q. Gut microbial metabolite butyrate improves anticancer therapy by regulating intracellular calcium homeostasis. Hepatology. 2023;78:88-102.
197. McBrearty N, Arzumanyan A, Bichenkov E, Merali S, Merali C, Feitelson M. Short chain fatty acids delay the development of hepatocellular carcinoma in HBx transgenic mice. Neoplasia. 2021;23:529-38.
198. Yan F, Zhang Q, Shi K, et al. Gut microbiota dysbiosis with hepatitis B virus liver disease and association with immune response. Front Cell Infect Microbiol. 2023;13:1152987.
199. Huo R, Chen Y, Li J, et al. Altered gut microbiota composition and its potential association in patients with advanced hepatocellular carcinoma. Curr Oncol. 2023;30:1818-30.
200. DeFilipp Z, Peled JU, Li S, et al. Third-party fecal microbiota transplantation following allo-HCT reconstitutes microbiome diversity. Blood Adv. 2018;2:745-53.
201. Tanoue T, Morita S, Plichta DR, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019;565:600-5.
202. Oh B, Boyle F, Pavlakis N, et al. The gut microbiome and cancer immunotherapy: can we use the gut microbiome as a predictive biomarker for clinical response in cancer immunotherapy? Cancers. 2021;13:4824.
203. Wu H, Zheng X, Pan T, et al. Dynamic microbiome and metabolome analyses reveal the interaction between gut microbiota and anti-PD-1 based immunotherapy in hepatocellular carcinoma. Int J Cancer. 2022;151:1321-34.
204. Matsubara T, Tanaka N, Patterson AD, Cho JY, Krausz KW, Gonzalez FJ. Lithocholic acid disrupts phospholipid and sphingolipid homeostasis leading to cholestasis in mice. Hepatology. 2011;53:1282-93.
205. Varanasi SK, Chen D, Liu Y, et al. Bile acid synthesis impedes tumor-specific T cell responses during liver cancer. Science. 2025;387:192-201.
206. Kudo M. A new era in systemic therapy for hepatocellular carcinoma: atezolizumab plus bevacizumab combination therapy. Liver Cancer. 2020;9:119-37.
207. Liu ZL, Zhu LL, Liu JH, Pu ZY, Ruan ZP, Chen J. Vascular endothelial growth factor receptor-2 and its association with tumor immune regulatory gene expression in hepatocellular carcinoma. Aging. 2020;12:25172-88.
208. Liu D, Song T. Advances in neoadjuvant therapy for hepatocellular carcinoma. Holist Integ Oncol. 2025;4:173.
209. Chiang CL, Chiu KWH, Chan KSK, et al. Sequential transarterial chemoembolisation and stereotactic body radiotherapy followed by immunotherapy as conversion therapy for patients with locally advanced, unresectable hepatocellular carcinoma (START-FIT): a single-arm, phase 2 trial. Lancet Gastroenterol Hepatol. 2023;8:169-78.
210. Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology. 2018;68:723-50.
211. Kim KJ, Kim JH, Lee SJ, Lee EJ, Shin EC, Seong J. Radiation improves antitumor effect of immune checkpoint inhibitor in murine hepatocellular carcinoma model. Oncotarget. 2017;8:41242-55.
212. Kim HJ, Park S, Kim KJ, Seong J. Clinical significance of soluble programmed cell death ligand-1 (sPD-L1) in hepatocellular carcinoma patients treated with radiotherapy. Radiother Oncol. 2018;129:130-5.
213. Behm B, Di Fazio P, Michl P, et al. Additive antitumour response to the rabbit VX2 hepatoma by combined radio frequency ablation and toll like receptor 9 stimulation. Gut. 2016;65:134-43.
214. Xie C, Duffy AG, Mabry-Hrones D, et al. Tremelimumab in combination with microwave ablation in patients with refractory biliary tract cancer. Hepatology. 2019;69:2048-60.
215. Zhang X, Zhu F, Wen J, Pan Z, Zhu Y. Factors influencing the timing of second-line therapy for the treatment of advanced hepatocellular carcinoma: a systematic review and meta-analysis. Holist Integ Oncol. 2025;4:179.
216. Kimura T, Kato Y, Ozawa Y, et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018;109:3993-4002.
217. Xie F, Chen B, Yang X, et al. Efficacy of immune checkpoint inhibitors plus molecular targeted agents after the progression of lenvatinib for advanced hepatocellular carcinoma. Front Immunol. 2022;13:1052937.
218. Wu R, Xiong J, Zhou T, et al. Quercetin/anti-PD-1 antibody combination therapy regulates the gut microbiota, impacts macrophage immunity and reshapes the hepatocellular carcinoma tumor microenvironment. Front Biosci. 2023;28:327.
219. Conche C, Finkelmeier F, Pešić M, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72:1774-82.
220. Liu HY, Pedros C, Kong KF, Canonigo-Balancio AJ, Xue W, Altman A. Leveraging the Treg-intrinsic CTLA4-PKCη signaling pathway for cancer immunotherapy. J Immunother Cancer. 2021;9:e002792.
221. Qiu Y, Wu Z, Chen Y, et al. Nano ultrasound contrast agent for synergistic chemo-photothermal therapy and enhanced immunotherapy against liver cancer and metastasis. Adv Sci. 2023;10:e2300878.
222. Yuan Y, Chen Y, Huang C, et al. Efficacy and safety of radiotherapy combined with immunotherapy and targeted therapy versus immunotherapy plus targeted therapy alone in unresectable hepatocellular carcinoma: a retrospective study. Front Oncol. 2025;15:1643304.
223. Yang H, Cong T, Luo Y, Yang C, Ren J, Li X. Prognostic effect of sarcopenia in hepatocellular carcinoma patients targeted with interventional therapy combined with immunotherapy and targeted therapy. J Hepatocell Carcinoma. 2024;11:175-89.
224. Sharma R, Pillai A, Marron TU, et al. Patterns and outcomes of subsequent therapy after immune checkpoint inhibitor discontinuation in HCC. Hepatol Commun. 2022;6:1776-85.
225. Andreata F, Laura C, Ravà M, et al. Therapeutic potential of co-signaling receptor modulation in hepatitis B. Cell. 2024;187:4078-94.e21.
226. Li FJ, Zhang Y, Jin GX, Yao L, Wu DQ. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8+ T cell in HCC patients. Immunol Lett. 2013;150:116-22.
227. Shi J, Li G, Liu L, et al. Establishment and validation of exhausted CD8+ T cell feature as a prognostic model of HCC. Front Immunol. 2023;14:1166052.
228. Liu M, Li L, Cao L, et al. Targeted delivery of CCL3 reprograms macrophage antigen presentation and enhances the efficacy of immune checkpoint blockade therapy in hepatocellular carcinoma. J Immunother Cancer. 2025;13:e010947.
229. Yang F, Hilakivi-Clarke L, Shaha A, et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78:1602-24.
230. Huang Y, Xie Y, Zhang Y, et al. Single-cell transcriptome reveals the reprogramming of immune microenvironment during the transition from MASH to HCC. Mol Cancer. 2025;24:177.
231. Thimme R, Neumann-Haefelin C. T cells engineered to carry a high-affinity HBV-specific T cell receptor: a potent weapon against advanced HBV-related HCC. Gut. 2025;Epub ahead of print.
232. Zhu W, Zhang Z, Chen J, et al. A novel engineered IL-21 receptor arms T-cell receptor-engineered T cells (TCR-T cells) against hepatocellular carcinoma. Signal Transduct Target Ther. 2024;9:101.
233. Bae WK, Lee BC, Kim HJ, et al. A phase I study of locoregional high-dose autologous natural killer cell therapy with hepatic arterial infusion chemotherapy in patients with locally advanced hepatocellular carcinoma. Front Immunol. 2022;13:879452.
234. Cheng Y, Gong Y, Chen X, et al. Injectable adhesive hemostatic gel with tumor acidity neutralizer and neutrophil extracellular traps lyase for enhancing adoptive NK cell therapy prevents post-resection recurrence of hepatocellular carcinoma. Biomaterials. 2022;284:121506.
235. Hu X, Shui Y, Shimizu S, et al. Targeted immune cell therapy for hepatocellular carcinoma using expanded liver mononuclear cell-derived natural killer cells. Neoplasia. 2024;58:101061.
236. Chen QT, Zhang ZY, Huang QL, et al. HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat Metab. 2022;4:1306-21.
237. Huang X, Yang J, Hu Y, et al. Protective effects of GalNac-modified red blood cell-derived extracellular vesicles against liver diseases. Int J Nanomedicine. 2025;20:8993-9017.
238. Yang K, Wang X, Song C, et al. The role of lipid metabolic reprogramming in tumor microenvironment. Theranostics. 2023;13:1774-808.
239. Yao H, Ma S, Huang J, et al. Trojan-horse strategy targeting the gut-liver axis modulates gut microbiome and reshapes microenvironment for orthotopic hepatocellular carcinoma therapy. Adv Sci. 2024;11:e2310002.
240. Li Z, Zhang Y, Hong W, et al. Gut microbiota modulate radiotherapy-associated antitumor immune responses against hepatocellular carcinoma Via STING signaling. Gut Microbes. 2022;14:2119055.
241. Tan J, Fang J, Luo W, et al. Co-delivery of chemotherapy and anti-angiogenic lipid via DPPA-LNPs potentiates anti-PD-1 immunotherapy. Int J Nanomedicine. 2025;20:14057-73.
242. Xian F, Wu C, Zhang G, Xu G. Efficacy and safety of immune checkpoint inhibitors combined anti-angiogenic therapy in patients with unresectable hepatocellular carcinoma: a meta-analysis. Medicine. 2022;101:e31479.
243. Jin ZC, Chen JJ, Zhu XL, et al. ; CHANCE2201 Investigators. Immune checkpoint inhibitors and anti-vascular endothelial growth factor antibody/tyrosine kinase inhibitors with or without transarterial chemoembolization as first-line treatment for advanced hepatocellular carcinoma (CHANCE2201): a target trial emulation study. EClinicalMedicine. 2024;72:102622.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.