


Emerging treatments for systemic lupus erythematosus
 0
0 
Abstract
It is well known that the failure of multiple past clinical trials in systemic lupus erythematosus (SLE) is partly due to the clinical and immunological heterogeneity of the disease. Appropriate trial designs, endpoints, and patient populations are required to assess the efficacy of SLE treatments. At the same time, interdisciplinary research is gradually revealing multiple targets that contribute to the pathophysiology of SLE. Currently, molecular-targeted therapies tailored to the disease are being developed as treatment strategies that do not rely on glucocorticoids. With the introduction of new treatments and technologies, clinical trials for SLE have significantly accelerated over the past decade. More than 300 interventional studies on SLE and lupus nephritis, both ongoing and planned, are underway. Various trials targeting different pathways are currently being conducted and SLE treatment strategies may be on the verge of a turning point in the near future. However, some challenges remain. In this review, we focus on promising agents in phase 2 clinical trials and beyond to highlight the latest perspectives and unresolved issues related to targeted therapies for SLE, as well as to explore future research directions and therapeutic strategies required for the implementation of personalized medicine for this rare disease.
Keywords
INTRODUCTION
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease characterized by the loss of self-tolerance, production of autoantibodies and antigen-antibody complexes, and involvement of multiple organ systems[1,2]. Although the incidence varies by ethnicity, it remains low, ranging from 1.5 to 7.4 per 100,000 person-years in Europe and 2.8 to 8.6 per 100,000 person-years in Asia[3]. Owing to its diverse clinical manifestations, diagnostic challenges, and limitations of current standard therapies, SLE is recognized globally as a rare condition with substantial unmet medical needs. Since 2000, numerous clinical trials have been conducted; however, challenges such as the need for patient stratification because of disease heterogeneity, unclear trial designs, and limited validated outcome measures have hindered progress[4]. Consequently, many drugs have failed to demonstrate superiority in clinical trials, thereby preventing their clinical application. However, since 2010, the development of molecular-targeted therapies and establishment of new clinical endpoints have made significant advances. Biologics targeting interferon (IFN) pathways and B cell-activating factor (BAFF) have enabled the achievement of clinical goals such as LLDAS (Lupus Low Disease Activity State) and DORIS (Definitions of Remission in SLE)[5,6]. Reflecting on these advances, the European League Against Rheumatism (EULAR) 2023 recommendations emphasize minimizing, and where possible discontinuing, glucocorticoid (GC) use[7].
Nevertheless, the currently approved agents anifrolumab and belimumab are effective in only 40%-70% of patients with SLE, highlighting a persistent unmet medical need. The development of precision medicine approaches (based on molecular characteristics and other factors) and the discovery of novel therapeutic agents are therefore warranted. Currently, over 300 clinical trials are investigating novel treatments for SLE, with more than 530 phase 2 or higher studies completed to date and 117 ongoing or planned trials. This review outlines the mechanisms and current developmental status of emerging therapeutic approaches, highlights key findings and remaining challenges from recent clinical trials, and discusses future research directions, clinical trial designs, and treatment strategies necessary for the implementation of personalized medicine for this rare disease. This section covers key drugs currently undergoing phase 2 or higher trials; trials that have completed less than or beyond phase 2, as well as small-scale clinical trials, are listed in the Supplementary Materials.
BIOLOGIC AGENTS
Biologic agents, which account for approximately 45% of the ongoing clinical trials, have undergone significant advancements owing to improvements in drug discovery technologies. These developments have resulted in biologics with longer half-lives, a reduced risk of adverse events, and enhanced functionality. Furthermore, the development of novel targeted therapies has been remarkable. This field is not limited to single-molecule targets; there is a growing exploration of agents targeting multiple molecules simultaneously, advancing the scope of biologics for SLE treatment. Table 1 and Figure 1 summarize the biologic agents targeting functional cellular molecules that are either currently undergoing clinical trials or planned to enter trials.
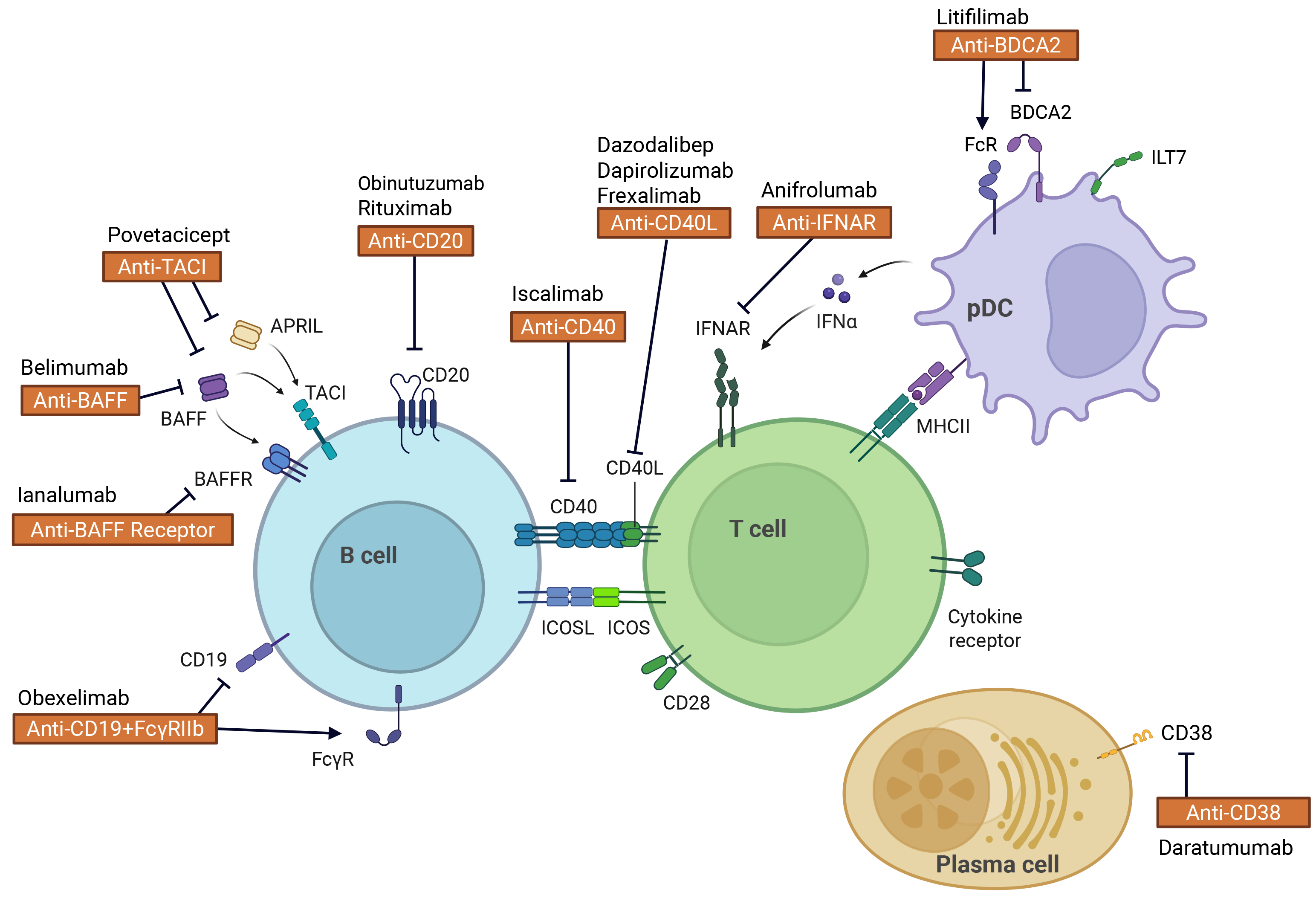
Figure 1. Therapeutic targets of biologics currently undergoing clinical trials for systemic lupus erythematosus. Therapeutic targets include molecules expressed on immune cells and cytokines produced by immune cells. BAFF: B-cell activating factor; CD40L: CD40 ligand; TACI: transmembrane activator and calcium-modulating cyclophilin ligand interactor; ICOS: inducible T-cell costimulator. ICOSL: ICOS ligand; IFNAR: type I interferon receptor. BDCA2: blood dendritic cell antigen 2; ILT7: immunoglobulin-like transcript 7; MHC II: major histocompatibility complex class II. Created in BioRender. Kanda, K. (2026) https://BioRender.com/q97c492.
Biologic agents targeting cells: phase 2 or higher clinical trials
| Target | Mechanism of action | Treatment | Trial phase | Status | Registration |
| B cells | Anti-CD19 antibody | Obexelimab | Phase 2 | Recruiting | NCT06559163 |
| Anti-CD20 antibody | Rituximab | Phase 4 | Recruiting | NCT05207358 | |
| Obinutuzumab | Phase 3 | Recruiting | NCT04702256 | ||
| Phase 2 | Recruiting | NCT05039619 | |||
| Phase 3 | Active, not recruiting | NCT04221477 | |||
| TACI antagonist | Telitacicept | Phase 2 | Recruiting | NCT05680480 | |
| Phase 3 | Recruiting | NCT05339217 | |||
| Phase 4 | Recruiting | NCT05666336 | |||
| Povetacivept | Phase 1/2 | Active, not recruiting | NCT05732402 | ||
| Anti-BAFF antibody | Belimumab | Phase 3 | Active, not recruiting | NCT03747159 | |
| Phase 4 | Recruiting | NCT06411249 | |||
| Phase 4 | Recruiting | NCT03543839 | |||
| Anti-BAFF receptor antibody | Ianalumab | Phase 2 | Active, not recruiting | NCT06293365 | |
| Phase 3 | Recruiting | NCT06133972 | |||
| Phase 3 | Recruiting | NCT05639114 | |||
| Phase 3 | Recruiting | NCT05624749 | |||
| Phase 3 | Recruiting | NCT05126277 | |||
| Anti-CD47/CD20 antibody | IMC-002 | Phase 1/2 | Not yet recruiting | NCT06535412 | |
| T cells costimulator | Anti-CD40L antibody | Frexalimab | Phase 2 | Recruiting | NCT05039840 |
| Dazodalibep | Phase 2 | Recruiting | NCT05201469 | ||
| Dapirolizumab Pegol | Phase 3 | Recruiting | NCT06617325 | ||
| Phase 3 | Enrolling by invitation | NCT04976322 | |||
| pDC | Anti-BDCA2 antibody | Litifilimab | Phase 2/3 | Recruiting | NCT05531565 |
| Phase 3 | Active, not recruiting | NCT04895241 | |||
| Phase 3 | Active, not recruiting | NCT04961567 | |||
| Phase 3 | Enrolling by invitation | NCT06044337 | |||
| Phase 3 | Enrolling by invitation | NCT05352919 | |||
| PC | Anti-CD38 antibody | Daratumumab | Phase 2 | Active, not recruiting | NCT04868838 |
B cell-targeted therapies
B cells play a critical role in the pathogenesis of SLE through effector functions, such as autoantigen presentation, autoantibody production, and T cell activation. In SLE, the survival and differentiation of autoreactive B cells are promoted by overproduction of BAFF and abnormalities in Toll-like receptor (TLR) function[8,9]. Since 2000, clinical trials have investigated agents such as rituximab, ocrelizumab, ofatumumab (targeting CD20), epratuzumab (targeting CD22), and atacicept (targeting BAFF/A proliferation-inducing ligand (APRIL)). However, these agents failed to achieve statistical significance in efficacy or were discontinued owing to adverse events, preventing their clinical application. Subsequent advancements, including better patient stratification, improved outcome measures, and enhanced antibody formulations, have enabled the development of multiple B cell-targeting agents. As of now, the EULAR 2023 recommendations endorse rituximab-based B-cell depletion therapy for refractory cases and those involving organ- or life-threatening conditions[7]. However, the criteria for these cases remain unclear. If ongoing trials yield favorable results, they could expand the therapeutic options for SLE treatment.
Anti-CD20 antibody
• Rituximab
Rituximab is a type I chimeric anti-CD20 monoclonal antibody, with previous small-scale trials and case reports demonstrating its efficacy in lupus nephritis (LN) and neuropsychiatric SLE (NPSLE)[10-12]. However, in clinical trials such as the EXPLORER trial (Phase 2/3, NCT00137969) for active SLE and the LUNAR trial (Phase 3, NCT02973789) for LN, rituximab failed to meet the statistical significance of the primary endpoints. Notably, though, reductions in severe relapses, anti-double-stranded DNA (dsDNA) antibody titers, and improvements in complement levels (C3 and C4) were observed in the rituximab group[13,14]. Failure to meet the primary endpoints, defined in the EXPLORER trial as the number of patients achieving major, partial, or no clinical responses according to British Isles Lupus Assessment Group (BILAG) at week 52, and in the LUNAR trial as the proportion of patients achieving complete (CRR), partial (PRR), or no renal responses (NRR) at week 52, has been attributed to several factors. These factors include the high efficacy of background immunosuppressive therapy in both studies, which may have masked additional drug effects; the use of stringent endpoint definitions, which may have underestimated partial clinical improvements; and, in the EXPLORER trial, the heterogeneity of SLE manifestations potentially affecting treatment response. Nevertheless, based on findings from real-world observational studies, small-scale clinical trials, and meta-analyses, rituximab is currently recommended as an induction therapy option for refractory LN in both the American College of Rheumatology (ACR) guidelines and European Alliance of Associations for Rheumatology (EULAR)/ The European Renal Association - European Dialysis and Transplant Association (ERA-EDTA) treatment recommendations[15,16]. Rituximab is approved for the treatment of refractory LN only in Japan. Currently, a phase 4 trial (NCT05207358: GLUREDLUP) is evaluating the safety and efficacy of rituximab for remission induction in proliferative LN, along with an observational study (NCT05659407: PREDICT) assessing BAFF and B cell phenotypes as biomarkers for response. In addition, a phase 4 trial (NCT05828147) is ongoing in patients with concurrent pulmonary hypertension. B-cell depletion with rituximab induced B-cell reconstitution and long-term remission in a subset of patients[17]. However, such outcomes are limited to a minority of cases, and the therapeutic effect is transient in most patients. Consequently, long-term control of disease activity remains challenging, underscoring the need for more potent and sustained B-cell-targeted therapies.
• Obinutuzumab
Obinutuzumab is a type II humanized anti-CD20 monoclonal antibody designed with low fucose expression in its fragment crystallizable (Fc) region, resulting in more than ten times the antibody-dependent cellular cytotoxicity (ADCC) and Antibody-Dependent Cellular Phagocytosis (ADCP) activity of rituximab. In SLE, a phase 2 trial involving 125 patients with LN was completed. The NOBILITY trial (NCT02550652) evaluated the efficacy of obinutuzumab versus placebo, each added to standard therapy consisting of GCs and mycophenolate mofetil (MMF). The primary endpoint was CRR at week 52. No statistically significant difference was observed between the placebo group (22 patients, 35%) and the obinutuzumab group (14 patients, 23%) (P = 0.115). However, at 104 weeks, the CRR was higher in the obinutuzumab group (obinutuzumab = 26 (41%) vs. placebo = 14 (23%) (P = 0.026). No increase in the incidence of severe adverse events or mortality was reported[18]. The limitations of this study included the small sample size, limited statistical power, stringent definition of the primary endpoint that excluded partial responses, and relatively short observation period, during which improvements were also seen in the placebo group, likely owing to the efficacy of background therapy. Nevertheless, the observation that drug efficacy could be demonstrated even under strict endpoint criteria during long-term follow-up provides a rationale for continued investigation of this agent. Phase 3 trials are currently underway for patients with LN and active SLE (NCT04702256: OBILUP, NCT05039619: POSTERITY, NCT04221477: REGENCY and NCT04963296: ALLEGORY). Compared with obexelimab and rituximab, obinutuzumab achieves more profound B-cell depletion, which may increase the risk of hypogammaglobulinemia and infection. However, obinutuzumab is receiving increasing attention owing to its potential to induce stronger immunological modulation and more durable disease control.
Anti-CD19 antibody
• Obexelimab
Obexelimab (XmAb5871) is a bispecific, non-cytolytic, humanized monoclonal antibody that binds to CD19 and Fc gamma (Fcγ) receptor IIb, inhibiting B cells, plasmablasts, and CD19-expressing plasma cells[19]. A randomized, double-blind, placebo-controlled phase 2 trial was conducted in 104 patients with SLE without life- or organ-threatening manifestations (NCT02725515). The primary endpoint was percentage of patients without loss of SLE disease activity improvement after 32 weeks. Although the primary endpoint did not achieve statistical significance, in the efficacy-evaluable population (patients who completed the study or discontinued owing to flares or treatment-related toxicity), the time to flare was significantly prolonged in the obexelimab group (P = 0.025). One limitation of this study was that only patients who experienced symptom relief with GC injections while discontinuing immunosuppressants other than hydroxychloroquine (HCQ) and who subsequently showed improvement in disease activity were included in the obexelimab or placebo groups. Transient disease control owing to GC use may have confounded the assessment, raising concerns about whether the actual efficacy of the studied drug was accurately captured. A potential bias in the composition of the efficacy-evaluable population cannot be excluded. A notable strength of this study was the incorporation of gene expression-based subgroup analyses, which enabled the identification of potential treatment-responsive subpopulations. Building on these findings, a multicenter, randomized, double-blind, placebo-controlled phase 2 trial (NCT06559163) is currently ongoing in patients with active SLE without LN, employing the BILAG-based Composite Lupus Assessment (BICLA) response as the primary endpoint. Unlike B-cell-depleting therapies, obexelimab does not deplete B cells and is, therefore, associated with a lower risk of infection and hypogammaglobulinemia. However, its onset of action appears to be slower than that of B-cell-depleting agents, and the risk of disease relapse may persist.
Anti-BAFF antibody
• Belimumab
Belimumab is a humanized monoclonal antibody targeting soluble BAFF. Soluble BAFF inhibits apoptosis of autoreactive B cells, class switch from immunoglobulin M (IgM) to immunoglobulin G (IgG) and differentiation to plasmablasts. It has been clinically used for over ten years to treat active SLE, providing significant benefits such as sustained reduction in disease activity, improvement in serological markers, and GC dose reduction[20]. Several clinical trials are currently underway to investigate the efficacy of belimumab in early SLE as part of the initial treatment (NCT06411249: BE-EARLY, NCT03543839) and multi-drug combination therapy strategies for severe LN (NCT05863936: BEAM). Previous studies demonstrated that the administration of belimumab along with MMF or cyclophosphamide during remission induction therapy for LN yields beneficial effects. An open-label extension of BLISS-LN (NCT01639339) demonstrated maintained efficacy and an acceptable safety profile[21,22]. However, treatment efficacy has been shown to be limited in patients with subendothelial deposits or a baseline protein/creatinine ratio greater than 3 g/g[23]. Additionally, the SynBioSe-1 study (NCT02284984), which investigated the concurrent administration of belimumab and rituximab, showed clinical responses. However, there are concerns regarding the re-proliferation of tissue-resident autoreactive B cells, recirculation of mature B cells, and elevated BAFF levels, suggesting that long-term B-cell depletion was not achieved. This may have contributed to an increased risk of relapse[24]. The ongoing SynBioSe-2 trial (NCT03747159), a multicenter randomized controlled phase 3 study, aims to evaluate the long-term clinical and therapeutic efficacy of B cell-targeting combination therapy using standard treatment followed by belimumab and rituximab. Additionally, a small randomized controlled trial (BEAT-LUPUS) comparing the combination of rituximab and belimumab versus rituximab plus placebo demonstrated a reduction in anti-dsDNA levels and a lower risk of severe flares in the combination group[25]. However, in refractory LN, a small-scale, multicenter, randomized, open-label clinical trial (NCT02260934: CALIBRATE) showed no improvement in clinical efficacy in terms of complete or partial renal failure when belimumab or placebo was administered after rituximab, cyclophosphamide, and GC therapy[26]. However, this was a small-scale study, and the long-term clinical efficacy has not been fully investigated. In this regard, the results of the SynBioSe-2 trial are awaited.
Anti-BAFF receptor antibody
• Ianalumab
Ianalumab (VAY736) is a deglycosylated human IgG1 monoclonal antibody targeting the BAFF receptor (BAFF-R). It depletes B cells through ADCC and blocks BAFF: BAFF-R signaling, which is crucial for B cell activation. A phase 2b, multicenter, randomized, parallel-group, double-blind, placebo-controlled trial (NCT03656562) conducted in 67 patients with active SLE evaluated the primary endpoint of SLE Responder Index 4 (SRI-4) response at 28 weeks, along with a composite measure of sustained GC reduction. The results showed that 44.1% (n = 15/34) of patients in the ianalumab (VAY736) group achieved the primary endpoint compared with 9.1% (n = 3/33) in the placebo group. At 52 weeks, patients who switched from active treatment to open-label ianalumab or from placebo to ianalumab showed continued effectiveness, with 45.5% (n = 15/33) and 40.6% (n = 13/32) achieving the target, respectively[27]. Although the primary endpoint evaluation period in this study was relatively short at 28 weeks, limiting the assessment of the long-term effects of ianalumab, a sub-analysis of circulating immune cell subsets demonstrated a statistically significant reduction in CD19-positive B cells by week 28 post-administration. Transitional, naïve, and memory B cells, as well as plasmablasts and plasma cells, were effectively depleted. Transcriptome analysis of whole blood samples revealed a significant downregulation of B cell-related genes, correlating with the observed decrease in B cell subsets, as measured by flow cytometry. Notably, reductions in neutrophil counts within the normal range and IFN gene signatures (IFN-GS) were observed in SRI-4 responders at week 28, suggesting that these parameters may serve as predictors of the response to ianalumab[28]. Currently, two phase 3 trials are recruiting participants, SIRIUS-SLE-1 (NCT05639114) and SIRIUS-SLE-2 (NCT05624749), which are investigating the addition of ianalumab to the standard treatment for SLE, focusing on its efficacy and safety. Additionally, the SIRIUS-LN trial (NCT05126277) will focus on evaluating the efficacy, safety, and tolerability of ianalumab when added to standard of care in LN. The depletion of memory B cells and plasmablasts distinguishes this agent from the existing BAFF inhibitor, belimumab. However, despite the availability of data from large-scale clinical trials, further investigation is required to fully elucidate the clinical efficacy, safety profile, long-term effectiveness, and how its indications differ from those of other BAFF signaling inhibitors.
TACI-targeting
• Telitacicept
Telitacicept is a fusion protein comprising the extracellular B lymphocyte stimulator (BLyS)/APRIL-binding domain of the transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) receptor linked to the human IgG Fc region. Unlike atacicept, telitacicept lacks the proprotein convertase cleavage site within TACI, rendering it more stable. Additionally, the stalk region of TACI, which exhibits strong affinity for BLyS and APRIL, is more highly conserved in telitacicept compared to atacicept. Telitacicept inhibits the differentiation of mature B cells into plasma cells and promotes apoptosis of long-lived plasma cells. In a previous phase 3 trial of atacicept (NCT00624338), two deaths occurred due to pulmonary infections complicated by alveolar hemorrhage, leading to early termination of the study[29]. Conversely, the ADDRESS II trial (NCT01972568), which implemented mitigation strategies such as mandatory vaccination and close monitoring of IgG levels, demonstrated no comparable safety concerns and showed efficacy versus placebo, suggesting the therapeutic potential of BLyS/APRIL inhibition. Against this background, a phase 2b trial of telitacicept (NCT02885610) conducted in China reported significantly higher rates of SRI-4 response across all dose groups, with an acceptable safety profile[30]. However, as this was a single-country study, limitations related to background treatment confounding and study design remain. In the United States, a phase 3 trial (NCT05306574: REMESLE-1) was being conducted to evaluate SRI-4 achievement at 24 weeks in patients with moderate to severely active SLE, but has since been discontinued.
• Povetacicept
Povetacicept (ALPN-303) is a TACI-Fc variant that binds to both APRIL and BAFF, and exhibits an IC50 (half-maximal (50%) inhibitory concentration) for APRIL and BAFF inhibition that is more than 50 times stronger than that of telitacicept. In preclinical studies, povetacicept showed the strongest inhibition of antibody responses and plasma cell formation compared to anti-BAFF antibodies and wild-type TACI-Fc. In the New Zealand Black (NZB)/New Zealand White (NZW) lupus model, povetacicept significantly suppressed anti-dsDNA autoantibodies and glomerulonephritis (including proteinuria and renal immune complex deposition) compared to an Fc control[31]. A phase 1/2 trial (NCT05732402: RUBY-3) is currently underway to assess the safety and efficacy of povetacicept in autoimmune kidney diseases including LN and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Although stronger disease control is anticipated, concerns remain regarding immunodeficiency and increased infection risk associated with excessive B-cell depletion. Moreover, as this was a small-scale, open-label study that included patients with LN and other manifestations, heterogeneity in disease pathology, variations in baseline treatments, differences in natural disease course, and potential evaluator bias cannot be excluded. Therefore, further investigation is required to conclusively verify efficacy.
Anti-CD40 antibody
• Iscalimab
Iscalimab (CFZ533) is an Fc-silent anti-CD40 antibody designed to inhibit the binding of immunoglobulins to Fc receptors (FcR), preventing ADCC and complement-dependent cytotoxicity (CDC). This design was implemented as a countermeasure to previous clinical trials in which CD40 antibodies mediated thrombosis via FcR engagement. In the NCT03610516 trial targeting LN, the primary safety endpoint was tolerable; however, no statistical significance was found for one of the primary endpoints, namely, changes in urine protein-to-creatinine ratio (UPCR). Although no significant difference was observed in the changes in the urinary protein-to-creatinine ratio (UPCR), a measure likely to reflect partial improvement, this finding should be interpreted with caution because of the small sample size and limited observation period. The efficacy of this drug in treating the underlying pathology of LN remains uncertain. Additionally, a phase II trial in non-renal SLE (NCT03656562) has been completed; however, no peer-reviewed publication providing formal efficacy evaluation is currently available.
Various novel B-cell-targeting therapies, including anti-B-cell antibodies and dual BLyS/APRIL inhibitors, are currently under investigation for SLE; however, several challenges remain. One issue with the depth of B-cell ablative therapy is whether it is possible to control B cells in the bone marrow and secondary lymphoid tissues, as well as autoreactive memory B cells and long-lived plasma cells, particularly those in the plasma cell compartment. However, detailed investigations in clinical trials have limitations. The second issue concerns the durability of B-cell ablative therapy. A paradoxical phenomenon has been reported, in which immune reconstitution following rituximab therapy increases blood BAFF levels, thereby promoting the differentiation of autoreactive B cells. Rituximab therapy has been associated with a high relapse rate and failure to achieve sustained remission in some cases. Therefore, it is necessary to determine the results of the new drugs currently being tested, as well as the combination therapy of rituximab and belimumab, in this regard. However, this requires a sufficient number of cases and a long-term observation period, and it is unclear whether a conclusion can be reached within the limited time of a clinical trial. Finally, there is the issue of safety, including the risk of infection that may be caused by short-term and long-term B cell removal or inactivation, as well as the breakdown of interactions with immune cells, such as other T cells and innate immune cells.
B-cell control in SLE is a key method for improving the pathology. In the future, establishing a "stepwise B-cell control strategy" to control the B-cell subset "stepwise and selectively", maximizing the use of new drugs, and close immune monitoring may be key to the success of B-cell targeting therapy.
T cell-targeted therapies-costimulatory targets
CD4+ T cells play a central role in immune responses and are classified into major subsets, including T helper (Th) 1, Th2, Th17, regulatory T (Treg), and follicular helper T (Tfh) cells[32]. These subsets exhibit plasticity and maintain physiological balance while being influenced by environmental factors such as cytokines. In patients with clinically manifest SLE, various cytokine imbalances and excessive activation, differentiation, and proliferation of CD4+ T cell subsets occur in the peripheral blood and affected tissues, correlating with disease activity and organ damage. Previous clinical trials targeting T cells have shown limited success. Contributing factors include the compensatory activation of alternative T cell subsets following the suppression of specific pathways, resulting in insufficient clinical efficacy, as well as organ-specific differences in T cell subsets that drive disease pathology. Multiple clinical trials are currently underway to target members of the Tumor Necrosis Factor (TNF) superfamily (e.g., CD40 ligand (CD40L)) and the CD28 immunoglobulin superfamily, including co-inhibitory molecules such as Programmed cell death protein 1 (PD-1) and Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and costimulatory molecules such as CD28, inducible T-cell costimulators (ICOS), and CD278, which are broadly expressed across various T-cell subsets.
TNF superfamily: CD40-CD40L target
The CD40-CD40L pathway mediates the interactions between dendritic (DCs) and T cells, as well as between T and B cells, and is involved in germinal center formation. This pathway plays a central role in the pathogenesis of SLE, production of autoantibodies, and their deposition in the kidneys. In particular, in patients with SLE and diffuse proliferative glomerulonephritis, glomerular and tubular epithelial cells exhibit high CD40 expression, which interacts with infiltrating CD4+ T cells to induce inflammation. Furthermore, interactions between CD40L on T cells and CD40 on B cells within the renal interstitium serve as sites for the local proliferation of naïve B cells and autoantibody-producing B cells in LN[33]. Several clinical trials using first-generation anti-CD40L antibodies have been conducted; however, these trials were discontinued because of reports of thromboembolic events. These events may result from soluble CD40L-anti-CD40L antibody immune complexes that trigger platelet activation and aggregation. Second-generation anti-CD40L and anti-CD40 antibodies have been developed as inactive or Fc-deficient formulations that reduce platelet activation, while maintaining full target binding. Multiple clinical trials are currently underway to test these new formulations.
• Dapirolizumab Pegol
Dapirolizumab Pegol (DZP) is a polyethylene glycol-conjugated, antigen-binding Fab fragment that targets CD40L. The lack of an Fc region minimizes its effects on platelet activation and aggregation. To date, two clinical trials in SLE have been conducted. In a phase 2b trial involving 182 patients with moderate to severe SLE (NCT02804763), the dose-response relationship between drug exposure and the primary endpoint - the proportion of patients achieving a BICLA response at week 24 - was evaluated. Although the BICLA response rate did not fit the prespecified dose-response model (P = 0.07), improvements in disease activity and immunological markers were observed, and DZP was well tolerated[34]. Post hoc analyses demonstrated reduced expression of multiple gene sets involved in B-cell activation and immunoglobulin production, as well as suppression of the type I IFN signature[35]. In this study, background therapy and endpoint design and the short observation period may have contributed to an underestimation of clinical efficacy; however, post hoc analyses suggested that treatment effects tended to emerge after week 28. A phase 3 trial (NCT04294667: PHOENYCS GO) involving 321 patients with moderate-to-severe SLE was completed. The primary endpoint was achieved, and at week 48, 49.5% (103/208) of patients in the DZP achieved a BICLA response, compared with 34.6% (37/107) in the Placebo (P = 0.0110; difference 14.6%). Furthermore, DZP was significantly more effective in achieving SRI-4 and reducing GC doses[36]. Currently, recruitment is ongoing for another phase 3 trial (NCT06617325: PHOENYCS FLY), a multicenter, randomized, double-blind, placebo-controlled parallel-group study evaluating the efficacy and safety of DZP in active moderate-to-severe SLE. The observation period will be extended to 48 weeks to evaluate achievement of BICLA. In addition, a phase 3 trial (NCT04976322) is planned to assess the long-term safety and tolerability of this drug.
• Dazodalibep
Dazodalibep (VIB4920) is an anti-CD40L-Tn3 fusion protein that lacks an Fc region, thereby eliminating the thromboembolic risk. Tn3 is a binding molecule designed by modifying the third fibronectin type III domain (FNIII-3) of human tenascin C. The Tn3 module is specifically designed to bind human CD40L and inhibit its interaction with the human CD40 receptor, thereby suppressing biological activity[37]. Currently, a multicenter, double-blind, placebo-controlled phase 2 clinical trial is recruiting patients with LN diagnosed with active nephritis. The primary endpoint of this trial is CRR at week 36, and the efficacy of dazodalibep or placebo added to the standard treatment with MMF and GC is evaluated (NCT05201469: VIBRANT). This study included an observation period of up to 60 weeks, which allowed long-term evaluation. However, the results were limited by the small sample size and stringent primary endpoints. A CRR can be achieved over time even with standard therapy, which may make it challenging to detect differences in treatment efficacy.
• Frexalimab
Frexalimab (SAR441344; INX-021) is a second-generation anti-CD40L monoclonal antibody designed with a modified Fc region that has low FcγRIIa binding, thereby eliminating the thromboembolic risk mediated by Fc. By inhibiting the interaction between CD40L and CD40, frexalimab is expected to suppress the differentiation of autoreactive B cells, thereby contributing to the management of SLE. Another notable feature of frexalimab is its ability to modulate T, B, natural killer (NK), and DC populations without inducing lymphocyte depletion, thereby enabling rapid immune reconstitution. Currently, a multicenter, randomized, placebo-controlled, double-blind phase 2 trial (NCT05039840: APATURA) is recruiting 116 patients with active SLE to evaluate its efficacy, with the primary endpoint being the SRI-4 response at week 24. However, in light of the results from the phase 2b trial of dapirolizumab, concerns remain about whether the sample size and observation period are sufficient to fully assess therapeutic efficacy. Based on the outcomes of this trial, the frexalimab trial may require refined strategies for patient stratification.
CD28 immunoglobulin superfamily: ICOS-ICOSL/CD28-CD80/86 target
ICOS and its ligand (ICOSL) represent another critical costimulatory molecule pair involved in T cell-dependent humoral immune responses. ICOS (CD278) is a member of the CD28 family. It is induced following T cell receptor (TCR) and/or CD28 costimulation, promoting T cell activation and proliferation. Notably, ICOS is highly expressed in Tfh cells and is essential for their generation, maintenance, and follicular homing, facilitating antibody affinity maturation within germinal centers. In patients with SLE, ICOS expression is upregulated and correlates with the expansion of circulating Tfh-like cells, enhanced IFN-γ expression, and the promotion of autoantibody production. Clinical trials using the human anti-ICOSL antibody prezalumab (AMG557) in SLE (NCT02391259, NCT00774943) have demonstrated that ICOSL blockade reduces anti-KLH IgG responses in patients with SLE. Thus, the ICOS/ICOSL axis represents a promising therapeutic target, particularly in autoimmune diseases involving autoantibodies.
• Acazicolcept
Acazicolcept (ALPN-101) is an Fc fusion protein with a humanized, mutated ICOS ligand (ICOSL) domain capable of simultaneously inhibiting the CD28 and ICOS pathways. In vitro studies have evaluated acazicolcept using peripheral PBMCs derived from healthy donors or patients with SLE. PBMCs were stimulated in the presence of acazicolcept, abatacept, or prezalumab, along with artificial antigen-presenting cells expressing CD80, CD86, ICOSL, and anti-CD3 (OKT3). Cytokine production and changes in gene expression were analyzed. The results demonstrated that acazicolcept, either alone or in combination, suppressed the expression of T-cell activation-associated genes more robustly than abatacept or prezalumab. Furthermore, acazicolcept inhibited the production of proinflammatory cytokines and suppressed immune activation pathways related to Th1 and Th2 activation and interleukin (IL)-17 signaling. In lupus model mice, acazicolcept significantly reduced the serum titres of anti-dsDNA antibodies and decreased IgG deposition in frozen kidney sections, which were not observed with abatacept monotherapy. These findings suggest that the simultaneous inhibition of both the ICOS and CD28 pathways may help regulate SLE-associated inflammation and autoimmune responses. A randomized, double-blind, placebo-controlled phase 2 trial (NCT04835441) evaluating ALPN-101 in moderate-to-severe active SLE has been completed; however, no peer-reviewed publication providing a formal evaluation of efficacy is currently available.
Although T cell-targeted therapies do not directly inhibit antibody production by B cells, they can indirectly regulate B cell function. Such therapies are expected to be effective in manifestations that are less dependent on B cells, such as cutaneous and articular involvement, and offer the advantage of reduced immunosuppression compared to B-cell-depleting therapies. These therapies may have a slower onset of action than B-cell-targeted approaches. Given the heterogeneity of T-cell subsets, identifying appropriate patient populations remains a challenge. The development of optimized treatment strategies, such as stepwise or combination approaches in which B-cell-targeted therapy is used for induction and T-cell-targeted treatment for maintenance, is warranted.
pDC-targeted therapies
Anti-BDCA2 antibody
• Litifilimab
Blood dendritic cell antigen 2 (BDCA2), a receptor specifically expressed on plasmacytoid DCs (pDCs), suppresses type I IFNs by pDCs. Litifilimab (BIIB059) is a humanized IgG1 monoclonal antibody that binds to BDCA2 and reduces signaling that leads to the production of type I IFNs, cytokines, and chemokines in endosome following its internalization. In a phase 1 trial (NCT02106897), litifilimab demonstrated potential benefits, particularly in patients with skin manifestations, by reducing IFN response gene expression, normalizing human myxovirus resistance protein 1 (MxA) expression, decreasing immune infiltration in skin lesions, and lowering Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI)-A scores[38]. In a phase 2 trial (NCT02847598, LILAC), the safety and efficacy of litifilimab were evaluated in two parts: Part A, targeting SLE with active skin symptoms and joint involvement, and Part B, targeting active cutaneous lupus erythematosus (CLE) with or without systemic symptoms. In Part A, which involved 102 patients, the efficacy of litifilimab was assessed. The original primary endpoint was the percent change from baseline in the CLASI-A score at week 12; however, in the revised protocol, this was changed to the change from baseline in the total active joint count - defined as the sum of swollen and tender joints - at week 24, based on a 28-joint assessment. The litifilimab group achieved a reduction of -15.0 ± 1.2, compared to -11.6 ± 1.3 in the placebo group (P = 0.04)[39]. Secondary endpoints evaluated included changes in CLASI, SRI-4, and Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K), but most of the secondary endpoints did not support the results of the analysis of the primary endpoint. Part B included 132 patients with active CLE and evaluated four dose levels. The primary endpoint, change in CLASI-A at week 16, showed significant improvements in the 50 mg, 150 mg, and 450 mg groups[40]. However, the short observation period limits conclusions regarding long-term efficacy and relapse prevention. Herpesvirus infections, including shingles, were observed in both parts, but infection risk related to pDC suppression may not have been fully captured.
Several phase 3 trials are currently ongoing. TOPAZ-1 (NCT04895241) is enrolling approximately 540 patients with active SLE, while TOPAZ-2 (NCT04961567) is enrolling about 480 patients with moderate to severe active CLE to evaluate SRI-4 at week 52. In addition, AMETHYST (NCT05531565) is targeting patients with CLE who are resistant or intolerant to antimalarial therapy, regardless of the presence of systemic manifestations.
Agents targeting pDCs have shown promise in treating SLE patients with cutaneous and articular involvement; however, not all patients exhibit a strong type I IFN signature, and the efficacy of pDC-targeted therapies may therefore be limited to specific subsets. Moreover, while pDC-derived type I IFN plays a critical role in early disease development, other pathways may predominate in the chronic phase, leaving unresolved challenges regarding long-term disease modification and safety.
ANTI-ILT7 ANTIBODY
• Daxdilimab
Daxdilimab (HZN-7734) is a human monoclonal antibody that targets immunoglobulin-like transcript 7, a pDC-specific cell surface protein, via ADCC. In a randomized, double-blind, placebo-controlled phase 2 trial (NCT04925934: RECAST SLE) conducted in 214 patients with moderate-to-severe active SLE, the efficacy of daxdilimab administered every 4 weeks (DAX Q4W) and every 12 weeks (DAX Q12W) was compared with that of placebo. The primary endpoints, including BICLA response at week 48 and achievement of oral GC doses of less than 7.5 mg or baseline levels, showed no significant differences between the DAX and placebo groups. However, the DAX Q4W group demonstrated a higher achievement of LLDAS than the placebo group, and positive trends were observed in the secondary and exploratory endpoints, suggesting potential therapeutic effects. A high placebo response rate was considered a contributing factor to failure to meet the primary endpoints. It was also taken into consideration that the strict endpoint of BICLA response + GC dose reduction had its limitations. A randomized, double-blind, placebo-controlled phase 2 trial evaluating the efficacy and safety of daxdilimab in patients with moderately to severely active discoid lupus erythematosus (DLE) (NCT05591222) was discontinued; similarly, a randomized, double-blind, placebo-controlled phase 2 trial in LN (NCT05540665) was also discontinued. There are currently no trials with Daxdilimab.
While drugs targeting pDCs show promise for treating SLE patients with cutaneous and articular manifestations, not all patients exhibit a strong type I IFN signature, and the efficacy of pDC-targeted therapies may be limited in certain subsets. Moreover, pDC-derived type I IFN plays a critical role in early disease pathogenesis, whereas other pathways may predominate in the chronic phase. Consequently, even in patients with a pronounced type I IFN signature, challenges remain regarding long-term disease modification and safety. In clinical practice, as with other therapeutic agents, the identification of predictive biomarkers - such as IFN-GS - is desirable to stratify patients likely to benefit from pDC-targeted treatment.
PC-targeted therapies
Anti-CD38 antibody
• Daratumumab
Daratumumab, a monoclonal antibody targeting CD38, which is highly expressed in plasma cells, has been shown to significantly reduce the number of malignant plasma cells in the bone marrow and has been approved for the treatment of multiple myeloma. Two case reports demonstrated the efficacy of daratumumab in refractory SLE. These reports indicated clinical and serological improvements following daratumumab administration. Additionally, daratumumab has been suggested to promote the reduction of autoreactive long-lived plasma cells, reduce type I IFN activity, and modulate effector T-cell responses[41]. In one of these cases, remission, according to the DORIS criteria, was maintained for three years after treatment, with normalized type I IFN activity throughout the observation period. Furthermore, the efficacy of daratumumab has been reported in six patients with refractory LN[42]. A phase II, single-center, open-label trial of daratumumab in patients with active LN (NCT04868838) is currently underway; however, the risk of bias is high owing to the small sample size and the open-label, single-center design.
Cytokine/cytokine receptor targeted therapies
In SLE, which is activated through the stimulation of innate immunity, leading to the activation of adaptive immunity, induction of autoreactive T cell development, and B cell activation and differentiation, cytokine signaling plays a crucial role in the differentiation, proliferation, and intercellular interactions of immune cells. A wide range of clinical trials targeting cytokines are underway, with the ongoing phase 2 or higher studies presented in Table 2.
Phase 2 or higher clinical trials targeting cytokines
| Target | Mechanism of action | Treatment | Trial phase | Status | Registration |
| Type 1 IFN | Anti-IFN-α receptor antibody | Anifrolumab | Phase 3 | Recruiting | NCT05138133 |
| Phase 3 | Active, not recruiting | NCT04877691 | |||
| Phase 3 | Recruiting | NCT06015737 | |||
| Phase 4 | Recruiting | NCT06594068 | |||
| Phase 3 | Recruiting | NCT05835310 | |||
| GR1603 | Phase 2 | Active, not recruiting | NCT06015230 | ||
| IL-2 | IL-2 mutant protein | Aldesleukin | Phase 3 | Recruiting | NCT05339217 |
Anti-IFNA receptor antibody
• Anifrolumab
Anifrolumab is a monoclonal antibody targeting the IFN receptor that inhibits all type I IFN signaling, including IFN-α, IFN-β, and IFN-ω. It has already been approved for the treatment of active SLE. Currently, multiple phase 2 or higher clinical trials are ongoing in LN, CLE, and pediatric SLE (NCT05138133, NCT04877691, NCT06015737, NCT06594068, NCT05835310, and NCT06659029). Among these, a large-scale randomized, double-blind, placebo-controlled phase 3 trial (NCT05138133: IRIS) in adult patients with active proliferative LN is promising. However, the primary endpoint is CRR at 52 weeks, and a key question remains as to whether a significant difference can be detected between CRR and placebo when combined with standard of care. Additionally, a randomized, double-blind, placebo-controlled phase 3 trial (NCT04877691: Tulip SC) is underway to assess the efficacy and safety of anifrolumab subcutaneous injection therapy for moderate-to-severe SLE. In the interim analysis, baseline demographic and clinical characteristics, as well as discontinuation rates, were well balanced between groups, and the primary endpoint was met, with a significantly higher proportion of patients achieving a BICLA response at week 52 in the anifrolumab group compared with placebo (59.4% vs. 43.9%, P = 0.0211)[43]. Other studies include a phase 3 randomized, double-blind, placebo-controlled trial (LAVENDER; NCT06015737) evaluated anifrolumab in adults with chronic and/or subacute CLE refractory or intolerant to antimalarial therapy, a registry assessing anifrolumab levels in breast milk and serum of lactating patients (NCT06594068) and a phase 3 trial examining the pharmacokinetics, safety, and efficacy of IV anifrolumab in paediatric patients aged 5-18 years with moderate-to-severe active SLE (BLOSSOM; NCT05835310).
Anifrolumab effectively suppresses type I IFN signaling in SLE and is being evaluated in phase 3 trials across multiple organ manifestations. However, its long-term renal effects, infection risk, and optimal timing of intervention remain unclear.
Anti-IFN-β antibody
• PF-06823859
Intracellular IFN-β in B cells is strongly correlated with anti-Sm antibodies, and the production of type I IFN by B cells has been implicated in SLE, particularly in patients who have previously experienced LN[44]. This process has been suggested to occur in transitional, naïve, and memory B cells. Histologically, high levels of B cell IFN-β are associated with immune complex deposition in the glomerular basement membrane and the anatomical features of both acute and chronic glomerular lesions. PF-06823859 (dazukibart) is a selective humanized IgG1 monoclonal antibody targeting IFN-β. A randomized, double-blind, placebo-controlled, multicenter phase 2 trial (NCT05879718) was conducted to evaluate the pharmacokinetics, safety, and efficacy of PF-06823859 in adult patients with active CLE or SLE with cutaneous manifestations; however, the trial was discontinued.
Anti-IFN-γ antibody
• Emapalumab
Emapalumab is a recombinant human IgG1 monoclonal antibody targeting human IFN-γ, approved for the treatment of primary hemophagocytic lymphohistiocytosis (HLH). An open-label, single-arm, multicenter study consisting of two cohorts (NCT05001737; EMERALD) has been completed. This study evaluated the efficacy, safety, tolerability, pharmacokinetics, and pharmacodynamics of emapalumab in pediatric and adult patients with secondary hemophagocytic lymphohistiocytosis/macrophage activation syndrome (sHLH/MAS) associated with Still’s disease - including systemic juvenile idiopathic arthritis and adult-onset Still’s disease - who had an inadequate response to high-dose glucocorticoid therapy. It also included patients with sHLH/MAS associated with SLE. The results of this study have not yet been published.
Currently, strategies targeting the IFN pathway using various agents directed at IFN-producing cells and type I IFN receptors are advancing. The range of inhibition of IFN signaling varies among these agents, necessitating tailored use based on organ involvement, with potential differences in safety profiles to be considered. Because type I IFN signaling is not involved in the pathogenesis of all SLE cases, a future challenge lies in maximizing therapeutic efficacy through more precise patient stratification.
INTRACELLULAR SIGNALING INHIBITORS
This section focuses on intracellular signaling inhibitors that are currently undergoing phase 2 or higher clinical trials, excluding biologic agents [Table 3]. The inhibitors shown here mainly include kinase inhibitors, TLR inhibitors, calcineurin inhibitors, and dual B-cell lymphoma 2 (BCL-2)/B-cell lymphoma extra-large (BCL-XL) inhibitors [Figures 2 and 3].
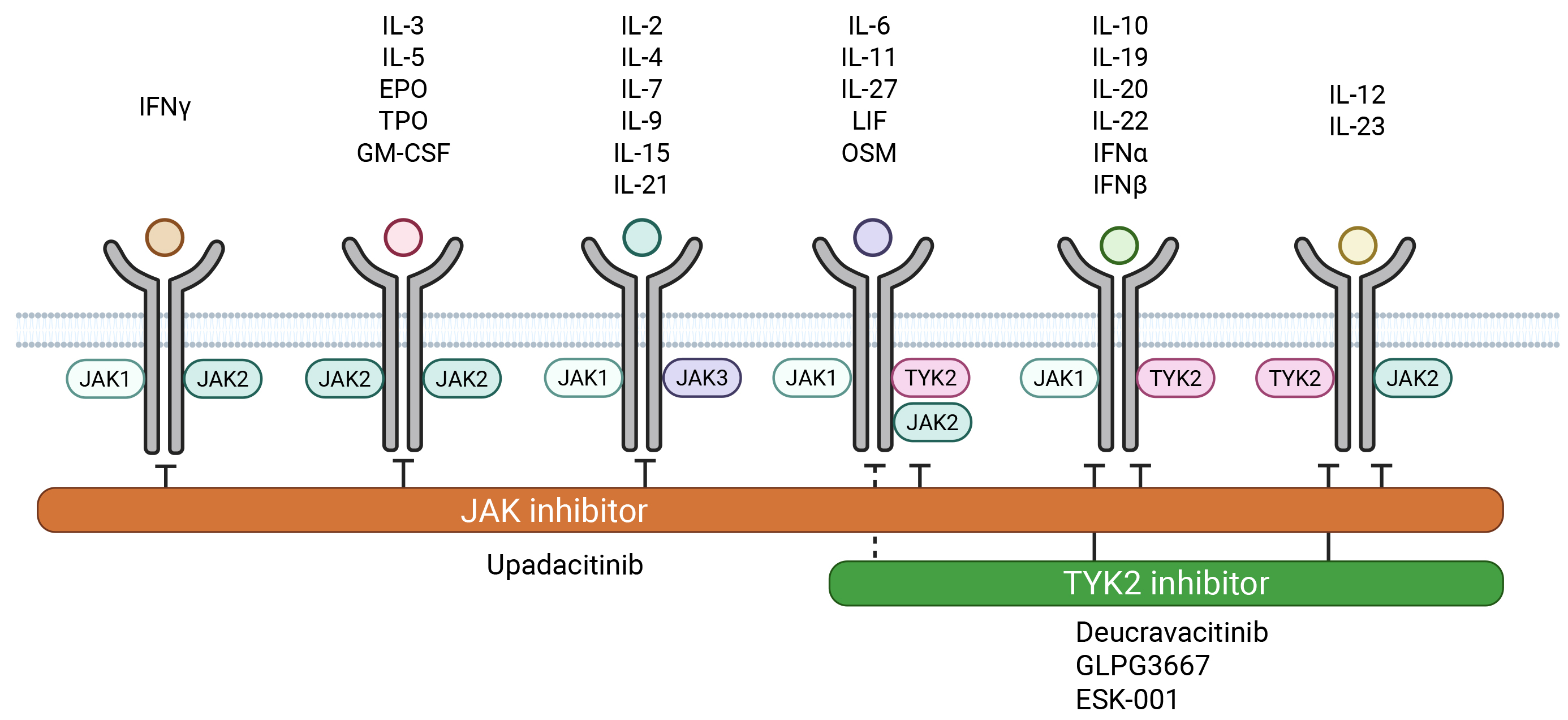
Figure 2. Cytokine signaling mediated by JAK/TYK2 inhibitors. Some cytokines share receptors and transmit signals through common pathways. TYK2 inhibitors have a strong affinity for inhibiting cytokines from the IL-12/23 and IL-10 families. JAK: Janus kinase; TYK2: tyrosine kinase 2. Created in BioRender. Kanda, K. (2025) https://BioRender.com/a34j761.
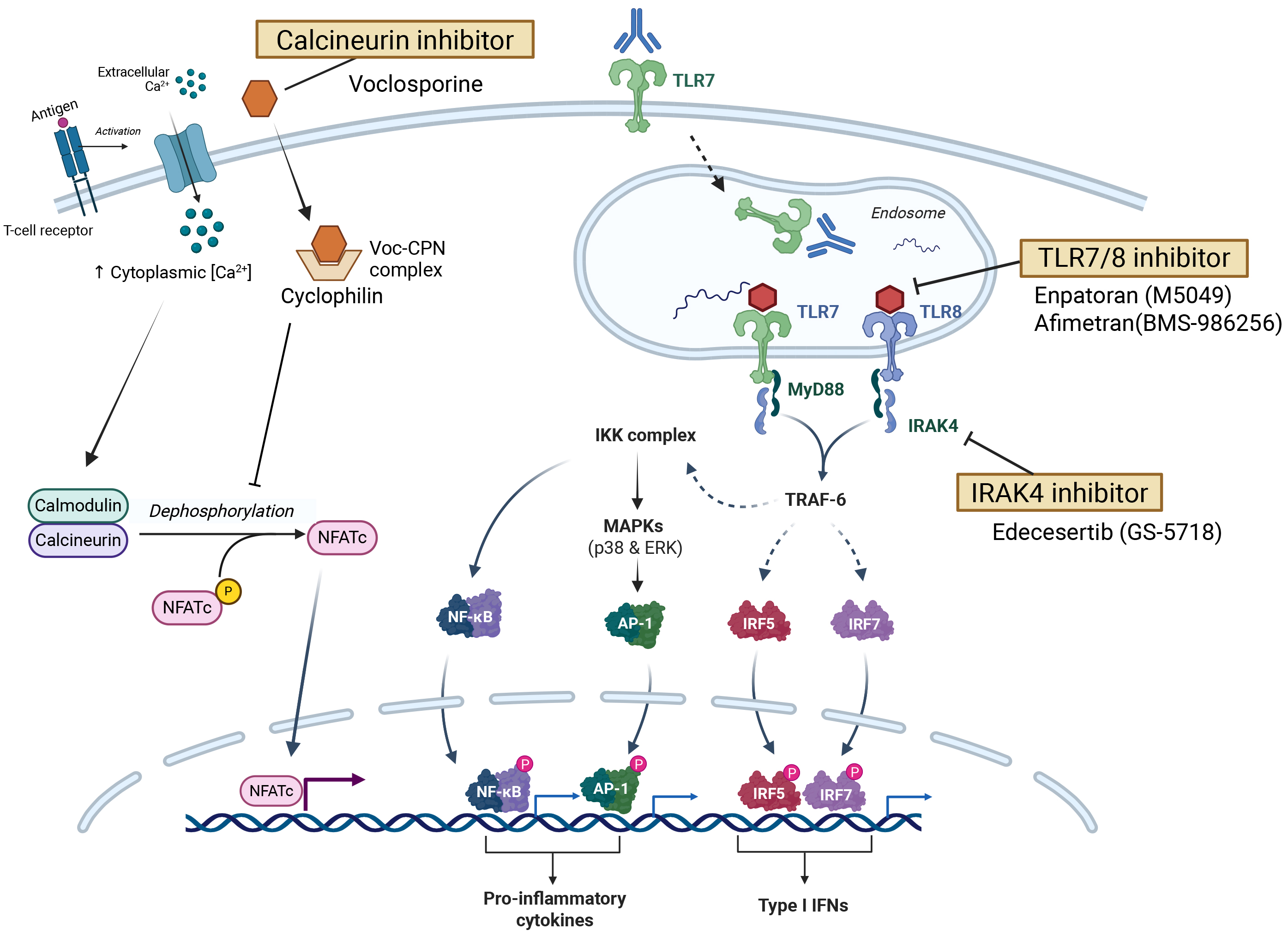
Figure 3. TLR-, IRAK4-, and calcineurin-mediated signaling and novel inhibitors. TLR: Toll-like receptor; IRAK4: interleukin-1 receptor-associated kinase 4; Voc: Voclosporine; CPN: cyclophilin. Created in BioRender. Kanda, K. (2026) https://BioRender.com/g84w088.
Phase 2 or higher clinical trials targeting intracellular signaling molecules
| Target | Mechanism of action | Treatment | Trial phase | Status | Registration | |
| Kinases | JAK/TYK2 | JAK inhibitor | Upadacitinib | Phase 3 | Recruiting | NCT05843643 |
| TYK2 inhibitor | Deucravacitinib | Phase 2 | Active, not recruiting | NCT04857034 | ||
| Phase 3 | Active, not recruiting | NCT05617677 | ||||
| Phase 3 | Active, not recruiting | NCT05620407 | ||||
| GLPG3667 | Phase 2 | Active, not recruiting | NCT05856448 | |||
| ESK-001 | Phase 2 | Active, not recruiting | NCT05966480 | |||
| BTK | BTK inhibitor | Orelabrutinib | Phase 2 | Recruiting | NCT05688696 | |
| Serine/threonine kinase | IRAK4 inhibitor | Edecesertib | Phase 2 | Active, not recruiting | NCT05629208 | |
| TLR | TLR7/8 | TLR7/8 inhibitor | Enpatoran | Phase 2 | Active, not recruiting | NCT05540327 |
| Afimetran | Phase 2 | Active, not recruiting | NCT04895696 | |||
| Calcineurin | Calcineurin | Calcineurin inhibitor | Voclosporin | Phase 3 | Recruiting | NCT06406205 |
| Phase 3 | Recruiting | NCT05288855 |
Janus kinase (JAK) inhibitors
Because multiple cytokines transmit signals through the shared Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway, JAK inhibitors differ from biologics in their ability to block a broad range of cytokine signals simultaneously[45]. Inhibition of cytokine signaling by JAK inhibitors can also affect the differentiation and proliferation of both innate and adaptive immune cells[46]. Over a decade has passed since their approval for rheumatoid arthritis (RA), and these inhibitors have demonstrated efficacy, even in cases resistant to multiple biologics, highlighting the broad impact of multi-cytokine blockade on the immune response. The therapeutic potential of JAK inhibitors in SLE has garnered significant interest, with several phase 3 or higher clinical trials being conducted to date. However, despite high expectations, practical clinical applications have not yet been achieved. Currently, the only ongoing phase 2 or higher trial is a phase 3 study evaluating upadacitinib.
• Upadacitinib
Upadacitinib is a JAK inhibitor with enhanced selectivity for JAK1 compared with other agents in its class. It has been approved for the treatment of various autoimmune diseases including RA. Clinical trials investigating upadacitinib have been designed to evaluate its safety and efficacy either as a monotherapy or in combination with the selective Bruton's tyrosine kinase (BTK) inhibitor elsubrutinib (ABBV-599). A phase 2 trial involving 341 patients with moderate-to-severe active SLE has been conducted (NCT03978520: SLEek)[47]. The primary endpoint was the proportion of patients achieving both an SRI-4 response and GC doses ≤ 10 mg/day by week 24. The results showed that the upadacitinib group (54%, P = 0.028) and the ABBV-599 high-dose group (48.5%, P = 0.081) had higher response rates than the placebo group (37.3%), with statistical significance observed for upadacitinib. Additionally, the 127 patients who completed the trial are enrolled in a long-term extension study lasting up to 104 weeks (NCT04451772)[48]. The proportion of patients achieving SRI-4 by week 48 increased to 85.4% in the ABBV-599 high-dose group, 82.1% in the upadacitinib 30 mg monotherapy group, and 61.3% in the placebo-to-ABBV-599 high-dose switch group. Across all groups, GC use was nearly eliminated by week 104 and safety remained acceptable.
Although JAK inhibitors are associated with concerns regarding venous thromboembolism and adverse cardiovascular events, no such events were reported in this trial cohort. Currently, a phase 3 trial (NCT05843643: SELECT-SLE) is recruiting patients with moderate-to-severe active SLE to assess the safety and efficacy of upadacitinib, with the primary endpoint being BICLA response at week 52.
In clinical trials conducted with tofacitinib and baricitinib, these drugs did not show statistical significance; however, important insights were obtained from post hoc analyses. The clinical trial of the pan-JAK inhibitor tofacitinib primarily focused on safety assessment and lacked sufficient statistical power to evaluate its efficacy. However, a post hoc analysis of NCT02535689 revealed that patients with the STAT4 risk allele, in particular, showed a reduction in the type I IFN signature and neutrophil extracellular traps (NETs), highlighting the importance of the IFN-JAK-STAT pathway in SLE treatment[49]. Although a phase 3 trial with baricitinib (NCT03616964, NCT03616912: SLE-BRAVE-II) failed, post hoc biomarker analysis using RNA and serum samples from phase 2 trials showed a significant reduction in the RNA expression of gene networks associated with SLE pathogenesis, including IFN, and a decrease in serum IL-12p40 and IL-6 levels[50], suggesting the potential of JAK inhibition in SLE. Furthermore, a phase 2 trial assessing the combination therapy of filgotinib/lanraplenib was discontinued because it failed to meet the primary endpoint (change in CLASI-A) (NCT03134222)[51].
JAK inhibitors have been associated with an increased risk of malignancy, cardiovascular events, and deep vein thrombosis in the ORAL Surveillance: A Postauthorization Safety Study of Tofacitinib in Comparison With TNF Inhibitors in Patients With Rheumatoid Arthritis, as well as an elevated risk of infections, such as herpes zoster. Many patients with SLE have antiphospholipid syndrome (APS) and thrombotic predisposition. Given that GC and other immunosuppressants remain the standard of care, safety concerns regarding JAK inhibitors persist. Nevertheless, emerging clinical trial data have begun to clarify the associations with responsive biomarkers, suggesting the potential for stratifying patients who are more likely to benefit from this therapy. The rapid onset of efficacy reported for symptoms that impact quality of life, such as cutaneous manifestations, arthritis, and fatigue, may offer a complementary option for symptoms refractory to existing immunosuppressive treatments.
Tyrosine kinase 2 (TYK2) inhibitor
Tyrosine kinase 2 (TYK2), a member of the JAK family, works together with JAK1 and JAK2 to mediate the signaling of key cytokines involved in SLE pathogenesis, including IL-12, IL-23, type I IFN, type III IFN, and IL-10[52]. TYK2 mutations are associated with an increased risk of inflammatory and autoimmune diseases. TYK2 is expressed in many immune cells, including T and B cells. When it is knocked out in B cells, TYK2 inhibits maturation and proliferation, and decreases globulin production. In T cells, TYK2 knockout suppresses Th1 differentiation and protects against autoimmune diseases. The development of selective TYK2 inhibitors has long been challenging, causing delays in their availability. However, with the development of selective TYK2 inhibitors and their pharmacological profiles, they have emerged as novel therapeutic targets in various autoimmune diseases. In recent years, it has become evident that highly selective TYK2 inhibitors do not interfere with IL-2 signaling, thereby preserving Treg cell differentiation. This suggests that the capacity to inhibit IFN signaling while maintaining Treg cells may offer a more targeted approach to ameliorating the pathology of SLE[53]. Currently, the inhibition of TYK2-mediated signal transduction is under investigation as a promising novel therapeutic strategy for SLE.
• Deucravacitinib
Deucravacitinib binds to TYK2 and blocks signaling through allosteric inhibition, demonstrating its high selectivity for inhibiting TYK2-related cytokines. The efficacy and safety of deucravacitinib in humans have been evaluated in clinical trials for various autoimmune diseases. In SLE, a phase 2 trial involving 363 patients (NCT03252587) showed that the primary endpoint of achieving SRI-4 at 32 weeks was significantly higher in patients receiving deucravacitinib (3 mg or 6 mg twice daily) than in those receiving placebo. In patients receiving deucravacitinib 3 mg twice daily, all secondary endpoints, including SRI-4, BICLA, LLDAS, and CLASI-50 responses, were achieved within 48 weeks[54]. Subsequent analysis of blood samples showed that deucravacitinib normalized the gene expression for type I IFN, IL-12, IL-23, and IL-10 signaling, with optimal results observed at a dose of 3 mg twice daily. However, higher doses did not provide additional benefits. Additionally, single-sample gene set enrichment analysis (ssGSEA) and cell deconvolution revealed increased Treg cells and decreased DCs in the deucravacitinib group, linking selective cytokine signaling inhibition to therapeutic efficacy.
Safety concerns have been raised about viral infections, particularly herpes zoster, caused by type I IFN inhibition. However, clinical trial results showed that although upper respiratory infections, acne, and skin rashes were observed more frequently in the deucravacitinib group, the incidence of serious adverse events was similar between the deucravacitinib and placebo groups. No deaths, opportunistic infections, tuberculosis, hematologic malignancies, major adverse cardiovascular events (MACE), or Venous Thromboembolism (VTE) were reported in any treatment group, and the incidence of herpes zoster did not increase. Currently, phase 3 trials are ongoing (NCT05617677: POETYK SLE-1 and NCT05620407: POETYK SLE-2). However, these trials excluded patients with significant organ involvement, such as those with severe LN requiring high-dose GCs and NPSLE, leaving the generalizability of efficacy to these populations for further investigation.
• GLPG3667
Cadefrecitinib (GLPG3667) has demonstrated efficacy in moderate-to-severe psoriasis in phase 2 trials and is currently undergoing a phase 2 trial (NCT05856448: GALACELA) for the treatment of active SLE. This trial evaluates the efficacy, safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG3667, with the primary endpoint being SRI-4 at 32 weeks. It is randomized, double-blind, and placebo-controlled, with ongoing recruitment. However, the evaluation period is short, and patients with active LN or NPSLE are excluded, limiting the study population. Therefore, challenges remain in establishing the clinical applicability of this treatment.
• ESK-001
Envudeucitinib (ESK-001) is currently being evaluated in a phase 2 randomized, double-blind, placebo-controlled trial in patients with active SLE (NCT05966480; LUMUS), with the primary endpoint being the BICLA response rate at 48 weeks. Notably, patients with active LN or NPSLE are excluded from the study population.
Therefore, TYK2 inhibitor therapy may be effective in patients with dominant type I IFN and IL-12/23 signaling pathways. Compared to JAK inhibitors, TYK2 inhibitors are more selective and are associated with a relatively lower risk of adverse effects. Concerns remain regarding the long-term safety, particularly the risk of infections, in the context of SLE, in which GCs and other immunosuppressive agents are standard therapies. Clinical trials have yielded results that support biomarker-based patient stratification, suggesting that TYK2 inhibitors may be a promising option for patients with cutaneous and articular SLE/CLE, especially those with elevated IFN and IL-12/23 activity.
Serine/Threonine kinase inhibitor
IRAK inhibitors
• Edecesertib (GS-5718)
Interleukin receptor-associated kinase (IRAK)4 is a serine/threonine kinase that regulates the production of inflammatory cytokines and type I IFNs downstream of TLRs and IL-1 receptors in immune cells. IRAK4 not only transphosphorylates and activates IRAK1, but also interacts as a scaffold protein with IRAK1, IRAK2, and Myeloid differentiation primary response gene 88 (MyD88) to form a signaling complex called the “Myddosome”. Myddosomes activate the nuclear factor kappa B (NF-κB) pathway, thereby promoting inflammatory cytokine production. TLR signaling (primarily through TLR7 and TLR9) is critical for SLE development, and IRAK4 inhibition is considered a promising target for lupus treatment[55]. Several kinase inhibitors with a single specificity for IRAK4 (IRAK4i) or dual specificity for IRAK1 and IRAK4 (IRAK1/4i) have been preclinically tested in animal models and in vitro, demonstrating good results in phase 1 trials, including edecesertib (GS-5718), zimlovisertib (PF-06650833), and R835. Currently, only edecesertib is being tested in phase 2 trials. A randomized, placebo-controlled, double-blind trial (NCT05629208) is currently being conducted to evaluate the efficacy, safety, and tolerability of edecesertib in patients with CLE, with the primary endpoint being CLASI-A response at 12 weeks. The trial was small in scale, and its statistical power was limited; thus, it was considered primarily as a proof-of-concept study.
IRAK4 acts downstream of the TLR/IL-1R signaling pathway; however, its function can be partially compensated by other signaling molecules such as IRAK1 and those involved in the MyD88-dependent pathway, which may limit clinical efficacy. IRAK4 not only possesses kinase activity but also functions as a scaffold protein that interacts with MyD88 and IRAK1/2. The extent to which IRAK4 inhibitors affect signal transduction via this scaffolding mechanism remains unclear.
Bruton's tyrosine kinase (BTK) inhibitor
BTK is involved in signal transduction, primarily in B cells, macrophages, and DCs, and regulates B cell activation and antibody production. BTK expression in peripheral blood B cells from patients with SLE is higher than that in healthy controls and has been confirmed to correlate with several parameters, including the SLEDAI, dsDNA antibodies, and proteinuria. To date, all clinical trials on BTK inhibitors for SLE have used second-generation inhibitors. These newer inhibitors, including evobrutinib (M2951) and fenebrutinib, exhibit improved selectivity and reduced off-target effects. Evobrutinib (M2951) is a covalent BTK inhibitor, and a phase 2 trial (NCT02975336) was conducted with SRI-4 achievement at 52 weeks as the primary endpoint. However, the trial did not achieve its primary endpoint. Fenebrutinib is a non-covalent BTK inhibitor that reversibly binds to BTK. In a phase 2 trial in patients with moderate-to-severe SLE (NCT03407482, NCT02908100), SRI-4 achievement at 48 weeks was the primary endpoint, but this trial also failed to meet the primary endpoint[56]. The lack of success of these trials may be attributed to several factors. First, BTK inhibitors do not affect T cell subsets or plasma cells that are involved in the pathogenesis of SLE and have low BTK expression. Additionally, BTK inhibitors do not affect type I IFN signaling, which plays a crucial role in SLE. Moreover, it is possible that the trials were not optimized in terms of appropriate patient selection, dosing schedules, or GC tapering criteria. Clinical trials of orelabrutinib and zanubrutinib were conducted; however, no formal results have been published to date.
• Orelabrutinib
Orelabrutinib (ICP-022) is a BTK inhibitor that exerts strong and sustained inhibitory effects by covalently binding to a specific cysteine residue (Cys481). In a phase 1b/2a trial (NCT04305197), dose-dependent achievement of SRI-4 was observed compared with placebo, with a particularly significant difference in SRI-4 responses in patients with high disease activity[57]. A randomized, double-blind, placebo-controlled phase 2b trial (NCT05688696) is being conducted to evaluate the efficacy and safety of orelabrutinib for SLE.
• Zanubrutinib
Zanubrutinib is a covalent BTK inhibitor. A randomized, double-blind, placebo-controlled phase 2 trial (NCT04643470) was conducted to assess its safety and efficacy in active proliferative LN, with complete renal remission as the primary endpoint. The trial was completed, but the results have not yet been published, and further detailed results are awaited.
BTK inhibitors enable broad immunomodulation by suppressing B-cells, monocytes, and DCs. However, they do not act on specific pathogenic cell populations such as T cells and long-lived plasma cells, making it difficult to completely inhibit autoantibody production. Since BTK is also expressed on platelets, there is a concern that its inhibition may increase the risk of bleeding. Although the ability to modulate multiple immune cell types via oral administration is appealing, clinical trial results to date suggest limitations to their practical application.
TLR inhibitors
TLRs are pattern recognition receptors activated by molecular signatures displayed by pathogens, such as bacteria and viruses, or by endogenous ligands released from cells that have undergone stress or damage. In humans, ten types of TLRs (TLR1-TLR10) have been identified, each structurally consisting of an extracellular domain that binds ligands and a cytoplasmic Toll/interleukin-1 receptor (TIR) homologous domain that regulates the intracellular signaling cascade. TLRs localize to either the cell surface or endosomal compartments that monitor cellular spaces. Endosomal TLRs recognize viral, bacterial, and self-derived nucleic acid fragments, including double-stranded RNA (dsRNA; TLR3), single-stranded RNA (ssRNA; TLR7 and TLR8), and single-stranded DNA (ssDNA; TLR9). TLR7 is expressed in B cells, monocytes, and pDCs, whereas TLR8 is predominantly expressed in neutrophils, monocytes, and myeloid DCs (mDCs). Although these two TLRs share common downstream signaling pathways, TLR7 activation predominates in the IFN regulatory factor (IRF) pathway, whereas TLR8 activation promotes NF-κB activation, which induces the production of inflammatory cytokines. Furthermore, activation of pDCs via TLR9 results in the production of type I IFNs, such as IFN-α and IFN-β, and drives the maturation of mDCs.
In the pathogenesis of SLE, B cells harbor genetic polymorphisms, and TLRs are thought to play a significant role as drivers of autoimmunity[58]. Because these TLRs are intracellular, drug development targeting them has historically been challenging. Multiple drugs are currently being developed for this purpose. With the elucidation of the structural requirements for effective ligand binding to TLR7 and the development of more suitable agents, clinical trials are currently underway for two small molecules targeting dual inhibition of TLR7 and TLR8: afimetoran (BMS-986256) and enpatoran (M5049).
TLR7/8 inhibitors
• Enpatoran
Enpatoran (M5049) is a quinoline derivative that functions as a dual inhibitor of TLR7/8. A randomized, double-blind, placebo-controlled, dose-ranging, parallel, and adaptive phase 2 trial (NCT05162586: WILLOW) was conducted to evaluate the efficacy and safety of enpatoran in SLE and CLE (SCLE and/or DLE) patients receiving standard treatment[59]. Cohort A included patients with CLE or SLE with active cutaneous involvement. In this cohort, enpatoran demonstrated a significant relationship in improvement of the CLASI-A score at week 16 and week 24. High rates of clinical response were observed, with up to 91.3% and 60.9% of enpatoran-treated patients achieving CLASI-50 and CLASI-70 responses, respectively, compared with 38.5% and 11.5% in the placebo group[60]. In Cohort B, 354 patients with moderate-to-severe SLE were enrolled in a randomized, double-blind, placebo-controlled study evaluating predefined dose levels. The primary endpoint was the dose-response relationship of BICLA response at week 24. Although a statistically significant overall dose-response was not observed, all enpatoran dose groups demonstrated numerically higher BICLA response rates than placebo, with the 25 mg twice-daily group achieving a significant improvement compared with placebo. Notably, the highest efficacy was observed in subgroups with high baseline IFN-GS or those receiving prednisone ≥ 10 mg/day. Type I IFN-GS was significantly reduced from week 2 across all enpatoran groups compared to the placebo group, with the reduction sustained through week 24. BICLA responders with GC tapering were observed in all enpatoran arms, and improvements in CLASI-A, as well as higher CLASI-50/70 response rates, were noted compared to the placebo group. Although infections have been reported, no apparent dose-dependent increase in incidence has been observed[61]. A long-term, double-blind, parallel-group phase 2 trial (NCT05540327: WILLOW LTE) is currently ongoing to evaluate the safety and efficacy of enpatoran for 48 weeks in participants from the WILLOW trial. The WILLOW trial supported the relevance of TLR7/8 inhibition in both SLE and CLE. The use of innovative trial designs and the evaluation of predictive markers of treatment response in this study represent significant advances; further investigations are warranted.
• Afimetoran
Afimetoran (BMS-986256) is an indole-based small molecule that functions as a dual inhibitor of TLR7 and TLR8. Preclinical studies using the NZB/NZW mouse model demonstrated a dose-dependent inhibitory effect on survival and kidney damage markers, including proteinuria, when administered alone or in combination with prednisolone. When combined with prednisolone, splenomegaly, age-associated B cells, and autoantibody production were suppressed. Currently, a phase 2, multicenter, randomized, double-blind, placebo-controlled trial (NCT04895696) is being conducted to evaluate the efficacy and safety of afimetoran in patients with active SLE.
• E6742
E6742 binds to a hydrophobic pocket in the inactive conformation of the ligand, maintaining TLR in its inactive form. During the development of E6742, the Japan Agency for Medical Research and Development (AMED) utilized its unique Medical Research and Development Innovation Base Creation (CiCLE) programme. A phase 1 trial in healthy adults (NCT04683185) and a phase 1/2 trial in patients with SLE (NCT05278663) have been conducted. In the phase 1/2 trial, 26 patients with SLE were assessed for the primary endpoint of E6742 safety and secondary endpoints, including pharmacokinetics, IFN-GS, and efficacy (BICLA response). The results showed that E6742 demonstrated a favorable safety profile, and its ability to suppress IFN-GS and efficacy in SLE was confirmed. These results provide the first clinical evidence supporting the use of E6742 in SLE treatment[62]. However, the study was limited by its small scale and short observation period, raising concerns regarding long-term efficacy. Since the trial focused on the cutaneous and articular forms of SLE, its relevance to severe organ manifestations such as LN or NPSLE remains limited.
TLR7 inhibitors
• DS-7011a
TLR7 is expressed not only in the endosomal compartment, but also on the cell surface, where it functions bidirectionally. When the anti-TLR7 monoclonal antibody preparation DS-7011a binds to TLR7, a TLR7/anti-TLR7 complex is formed. This complex is internalized into the endosomal compartment and, as it accumulates, TLR7 molecules are covered by the formulation, preventing the recognition of nucleic acids. Anti-TLR7 agents affect not only B cells, but also DCs, macrophages, and other cells, thereby inhibiting TLR7 responses. A phase 1/2 clinical trial of DS-7011a in patients with SLE (NCT05638802) has been completed, although the results have not yet been published. However, given that the pathogenesis of SLE involves TLR7 and other endosomal TLRs such as TLR8 and TLR9, selective inhibition of TLR7 alone may be insufficient to fully control disease activity.
In SLE, TLR inhibition therapy has already shown some proof-of-concept and targeting TLRs is considered one of the treatments that addresses the pathophysiology of the disease. This agent may serve as a therapeutic option, primarily for patients with mild-to-moderate cutaneous and articular SLE/CLE who are positive for biomarkers associated with high IFN activity. If future clinical trials yield favorable results, this therapy could be positioned as an adjunctive treatment for patients refractory to GCs or immunosuppressants. However, there are still concerns regarding the specificity of the targets, particularly the influence of TLRs other than TLR7, 8, and 9 on SLE inflammatory pathology and the risk of infection.
Calcineurin inhibitor
• Voclosporin
Calcineurin inhibitors suppress T-cell activation by inhibiting calcineurin-dependent dephosphorylation of nuclear factor of activated T-cells (NFAT), thereby reducing cytokine gene transcription. Conventional agents such as cyclosporine A and tacrolimus are effective but limited by toxicities, including nephrotoxicity and metabolic disturbances, necessitating careful monitoring. Voclosporin is a semi-synthetic cyclosporine derivative developed to improve pharmacokinetics and reduce metabolic adverse effects, without affecting mycophenolic acid exposure when combined with MMF for LN. In the phase 3 AURORA-1 trial (NCT03021499), voclosporin combined with standard therapy significantly improved complete renal remission rates at 52 weeks compared with placebo[63], and long-term safety was confirmed in AURORA-2 (NCT03597464), leading to its approval for LN. Ongoing phase 3 studies are further evaluating its efficacy and use in younger patients (NCT06406205, NCT05288855: VOCAL). However, voclosporin remains associated with calcineurin inhibitor-related adverse effects, such as reduced glomerular filtration rate and hypertension, and complete renal remission and GC withdrawal remain difficult to achieve in many patients.
Dual BCL-2 and BCL-XL inhibitor
• APG-2575
Lisaftoclax (APG-2575) is a dual inhibitor of B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma extra-large (Bcl-xL), two anti-apoptotic factors that prevent lymphocyte apoptosis. By selectively binding to these proteins, APG-2575 induces lymphocyte apoptosis. It has been approved for the treatment of chronic lymphocytic leukaemia and small lymphocytic lymphoma. Currently, a randomized, double-blind, placebo-controlled phase 1/2 trial (NCT06182969) is underway to evaluate the safety, pharmacokinetics, and pharmacodynamics of APG-2575 in patients with mild-to-moderate SLE. Lisaftoclax is a small-molecule drug that uniquely suppresses autoantibody production by controlling and eliminating autoreactive B cells, distinguishing it from other B-cell-targeted approaches. However, its clinical significance in SLE remains unclear.
Intracellular signaling inhibitors, including JAK, TYK2, TLR, calcineurin, and apoptosis-targeting agents, represent a rapidly evolving therapeutic class in SLE, offering oral, multi-cytokine modulation beyond biologics. Emerging data support their efficacy, particularly in cutaneous and articular disease, while highlighting the importance of biomarker-driven patient stratification. Ongoing trials will clarify their long-term safety, organ-specific applicability, and positioning within SLE treatment paradigms.
Miscellaneous
Clinical trials targeting the complement pathway and novel therapies focusing on selective sphingosine-1-phosphate (S1P1) receptors and neonatal Fc receptor (FcRn), among other innovative approaches, are currently underway [Table 4].
Phase 2 or higher clinical trials in miscellaneous treatments
| Target | Mechanism of action | Treatment | Trial phase | Status | Registration |
| Complement pathways | Bifunctional C5 antibody/Factor H fusion protein | KP-104 | Phase 2 | Not yet recruiting | NCT05504187 |
| Factor B inhibitor | Iptacopan | Phase 2 | Recruiting | NCT05268289 | |
| mTOR | mTOR signaling inhibitor | Sirolimus | Phase 2 | Active, not recruiting | NCT04582136 |
| Phase 2 | Not yet recruiting | NCT04736953 | |||
| Mesenchymal stem cells | Mesenchymal stem cells | Phase 2 | Active, not recruiting | NCT02633163 | |
| Phase 2 | Recruiting | NCT04835883 | |||
| Phase 2 | Recruiting | NCT03673748 | |||
| SGLT2 | SGLT2 inhibitor | Dapagliflozin | Phase 2 | recruiting | NCT06155604 |
| S1P1 receptor | S1P receptor 1 modulator | Cenerimod | Phase 3 | Recruiting | NCT05648500 |
| Phase 3 | Recruiting | NCT05672576 | |||
| Phase 3 | Enrolling by invitation | NCT06475742 | |||
| FcRn | FcRn inhibitor | Nipocalimab | Phase 2 | Not yet recruiting | NCT04883619 |
| Bcl-2/Bcl-xL | dual Bcl-2/Bcl-xL inhibitor | APG-2575 | Phase 1/2 | Recruiting | NCT06182969 |
COMPLEMENT PATHWAYS-TARGETED THERAPIES
The complement system plays a dual role, both promoting tissue damage through activation and providing protective effects such as the clearance of apoptotic cells and the suppression of IFN-α production. In SLE, the classical pathway is activated by immune complex formation, which leads to complement consumption and decreased serum complement levels. Complement abnormalities have been implicated particularly in LN, suggesting that complement activation may serve as a novel therapeutic target in patients with predominant complement activity. Currently, one clinical trial targeting the complement pathways is underway.
• Iptacopan
Iptacopan (LNP023), an inhibitor of complement factor B (FB) in the alternative complement pathway, blocks C3 convertase (C3bBb) formation. A phase 2, randomized, double-blind, placebo-controlled study (NCT05268289) is currently recruiting participants to evaluate the efficacy, safety, and tolerability of LNP023 in combination with standard treatment for patients with active LN. This large-scale, long-term trial will allow for the concomitant use of standard therapy and is expected to demonstrate GC-sparing effects and efficacy in treatment-resistant nephritis.
Novel agents targeting the complement pathway offer a new approach to SLE by directly modulating immune complex formation and complement activation. They may benefit patients refractory to conventional therapy, particularly those with LN, and support GC-sparing strategies. However, inhibition of complement raises concerns about bacterial infections, requiring careful patient selection and preventive measures such as vaccination.
NEONATAL FcRn TARGETED THERAPIES
FcRn plays a key role in regulating IgG and albumin metabolism and transport. It protects internalized IgG from lysosomal degradation through endocytosis, recycling it back into circulation. FcRn also facilitates the bidirectional transport of IgG across barriers such as the blood-brain barrier, making it a potential target for drug delivery systems in autoimmune diseases. The inhibition of FcRn is thought to suppress the recycling of pathogenic autoantibodies and improve drug delivery across biological barriers. In SLE, autoantibodies are thought to evade degradation and persist in the circulation for prolonged periods, contributing to immune complex formation, complement activation, and sustained organ damage. FcRn has been implicated in the uptake of immune complexes by macrophages, which may contribute to the promotion of inflammatory responses. The inhibition of FcRn is expected to shorten the half-life of total IgG, thereby reducing autoantibody levels and potentially mitigating immune complex formation and tissue deposition.
• Nipocalimab
Nipocalimab is a novel, high-affinity, fully human, non-glycosylated IgG1 monoclonal antibody that selectively inhibits FcRn without an effector function. A phase 2, randomized, placebo-controlled, double-blind, parallel-group study (NCT04882878) was conducted to evaluate the efficacy and safety of nipocalimab in active SLE, with the primary endpoint being the achievement of SRI-4 at 24 weeks. The target population was expected to primarily consist of patients with skin and joint manifestations without extensive organ involvement. The results are awaited to determine whether short-term assessment using the SELENA-SLEDAI can adequately evaluate organ damage, such as rash and arthritis. Although detailed results have not yet been published in a peer-reviewed format, sponsor-reported topline data indicate that the trial met its primary endpoint (SRI-4 response at Week 24) and key secondary endpoints, and that the safety profile was consistent with prior studies.
CELL-BASED THERAPY
CART therapy
Chimeric antigen receptor T-cell therapy (CART) involves genetically modifying T cells to express chimeric antigen receptors (CARs), which enable T cells to target and kill cells bearing specific antigens. This innovative treatment strategy has garnered attention because of its ability to activate a signaling cascade that enhances cytotoxic activity against target cells. While detailed discussions on CAR-T cell therapy are provided in other sections, it is noteworthy that ongoing trials targeting SLE predominantly use CAR-T cells engineered to recognise CD19 on B cells. In preclinical studies, CAR-T cells targeting CD19 showed superior effectiveness in depleting B cells compared to conventional B cell-depleting antibody therapies in SLE model mice[64]. Additionally, case reports of five patients with refractory active SLE treated with CAR-T therapy showed promising results[65,66]. All patients achieved remission according to the DORIS criteria within three months of treatment, and remission was maintained for a median of eight months during follow-up without additional therapy. Furthermore, circulating CAR-T cells proliferated until approximately day 9 post-infusion, after which their levels rapidly decreased. The re-emerging B cells were naïve B cells lacking class-switching B cell receptors, and no disease flares were observed during the follow-up period. In addition to CD19, various CAR constructs targeting antigens such as CD20 and BCMA have been developed, and clinical trials are currently underway [Table 5].
Phase 2 or higher clinical trials of CAR-T therapy
| Agent | Mechanism of action | Trial phase | Status | Registration |
| CAR-T | CD19-targeting CAR-T cell | Phase 1/2 | Recruiting | NCT06585514 |
| Phase 1/2 | Active, not recruiting | NCT05938725 | ||
| Phase 2 | Recruiting | NCT06581198 | ||
| Phase 1/2 | Recruiting | NCT06347718 | ||
| Phase 1/2 | Recruiting | NCT06106906 | ||
| Phase 1/2 | Active, not recruiting | NCT06342960 | ||
| Phase 1/2 | Recruiting | NCT06189157 | ||
| Phase 1/2 | Recruiting | NCT06121297 | ||
| Phase 1/2 | Active, not recruiting | NCT05798117 | ||
| Allogeneic CD19 CAR-γδ T cells | Phase 1/2 | Recruiting | NCT06106893 | |
| BCMA/CD19 CAR-T cells | Phase 1/2 | Recruiting | NCT06428188 | |
| Phase 1/2 | Recruiting | NCT06350110 | ||
| RNA-engineered CAR-T cell | Phase 2 | Recruiting | NCT06038474 |
Although large-scale trial results are not yet available, some patients achieved complete remission, suggesting that CAR-T cell therapy may represent a novel treatment strategy for refractory multidrug-resistant SLE. Successful discontinuation of GCs and immunosuppressants could potentially reduce the risk of complications, such as infections and osteoporosis. However, numerous challenges remain, including increased susceptibility to infections owing to intensive immunosuppression before CAR-T cell infusion, high costs, and possible effects on non-autoimmune regulatory mechanisms. In response to these challenges, “Bispecific T cell engager antibodies” that simultaneously target both T and B cells - achieving B-cell depletion comparable to CAR-T therapy while offering improved safety and easier clinical application - have attracted considerable attention.
T-cell engager
T-cell engager (TCE) engagers are bispecific antibodies with dual binding sites that target CD3 on T cells and antigens on target B cells. This dual specificity activates CD3+ T cells, leading to the destruction of the target cells. Although no phase 2 trials are currently underway, T cell engagers are attracting attention as alternatives to CAR-T cell therapy, addressing technical challenges and safety concerns by achieving comparable B-cell depletion while improving safety and clinical applicability.
RO7507062 is a Bispecific T-cell engager (BiTE) antibody targeting CD3 and CD19, which has demonstrated potent B-cell depletion in preclinical studies using PBMCs derived from patients with SLE[67]. Imvotamab (IGM-2323) is an IgM-based bispecific antibody that targets CD3 and CD20. Studies using human peripheral blood mononuclear cells from healthy donors and patients with autoimmune diseases, including SLE, RA, and multiple sclerosis, have confirmed that its efficacy in B-cell depletion is comparable to that of rituximab. Investigations on non-human primates have confirmed their ability to kill CD20-expressing target cells directly in tissues[68]. CLN-978 is a fusion protein composed of tandemly arranged single-chain variable fragments (scFvs) specific for CD19 and CD3 combined with a single-domain albumin binder that extends its serum half-life. CLN-978 exhibits a high affinity for CD19 and induces profound depletion of B cells in the peripheral blood, bone marrow, spleen, and lymphoid tissues[69]. Although currently limited to phase 1 clinical trials (NCT06613360), TCE is expected to emerge as a novel therapeutic strategy. While clinical evidence is currently limited to off-label use, teclistamab, a bispecific antibody targeting CD3 and B-cell maturation antigen (BCMA), has shown potential efficacy in severe cases of SLE. Teclistamab has been suggested to deplete long-lived plasma and memory B cells in patients with SLE[70].
Currently, the efficacy of TCE therapy for SLE remains unclear. TCE treatment in autoimmune diseases carries risks, such as unintended activation of autoreactive T cells, cytokine release syndrome, off-target effects, and damage to normal immune cells. Further clinical trials are necessary to thoroughly evaluate the efficacy and safety of this approach.
Haploidentical allogenic bone marrow-derived mesenchymal stem cell
Mesenchymal stem cells (MSCs), particularly those derived from the umbilical cord (hUC-MSCs), have shown promising potential in the treatment of autoimmune diseases such as SLE, owing to their immunomodulatory effects and tissue repair capabilities. These cells can suppress T cells, self-reactive B cells, and macrophages, and inhibit autoantibody production. In a phase 1/2 trial involving 40 patients with active SLE (NCT01741857), intravenous hUC-MSCs were safe and improved the SLEDAI and BILAG scores, although some patients relapsed after six months. A phase 2 trial involving 18 patients with LN (NCT01539902) did not show significant efficacy, leading to discontinuation. Ongoing phase 2 and 3 trials, including NCT02633163, NCT04835883, NCT03917797, and NCT03673748, aim to further evaluate MSC therapy in SLE and LN, with a phase 3 trial exploring low-dose IL-2 therapy combined with hUC-MSCs for LN (NCT05631717). These studies sought to determine the potential of MSCs for the treatment of autoimmune diseases, such as SLE. However, MSCs exhibit variability in their immunomodulatory capacity and proliferative potential depending on donor factors such as age, sex, and tissue of origin (e.g., bone marrow or umbilical cord). Genetic and functional changes can occur owing to passage number and culture conditions, raising concerns about quality deterioration and reduced immunosuppressive efficacy. These factors make it difficult to standardize the product quality across different trials. Multiple challenges related to safety and supply pose significant hurdles for clinical applications.
S1P1 RECEPTOR MODULATOR
Sphingosine-1-phosphate (S1P) receptor dysfunction in SLE is thought to contribute to the accumulation of self-reactive lymphocytes in tissues, the production of inflammatory cytokines such as IL-6 and IFN-α, and fibrosis in organs such as the kidneys. Normally, lymphocytes migrate from lymph nodes to the bloodstream following a gradient of S1P concentration (lymph nodes < blood), eventually reaching the sites of inflammation. S1P receptor modulators indirectly inhibit this migration, thereby preventing lymphocytes from reaching the inflamed tissues. Therefore, S1P receptors may serve as pharmacological targets for autoimmunity and inflammation. Fingolimod, a nonselective S1P receptor modulator, has shown efficacy in multiple sclerosis, and several clinical trials are currently investigating the use of other S1P receptor modulators, such as cenerimod, in SLE.
• Cenerimod
In a phase 1/2 trial of cenerimod (ACT-334441; NCT02472795), a dose-dependent decrease in lymphocyte counts, anti-dsDNA antibody levels, and disease activity was observed. A phase 2b trial in 427 moderate-to-severe SLE (NCT03742037) failed to meet its primary endpoint but showed improvements in disease activity, plasma IFN-α levels, and reductions in B and T cell counts in the 4-mg group[71]. One factor contributing to the failure to meet the primary endpoint in this trial may have been the difficulty in demonstrating statistical significance owing to the multiple comparison adjustments inherent in the multi-arm, multi-dose study design. The randomization design and heterogeneity of background therapies may have interfered with the accurate evaluation of treatment effects. To address these issues, further evaluation of cenerimod's efficacy and safety was performed using a single fixed dose, focusing on the IFN-1 high subgroup, assessing sustained endpoints (such as continuous response over four months), and implementing standardized background therapy criteria. Currently, randomized, double-blind, placebo-controlled phase 3 trials (OPUS-1, OPUS-2, and OPUS-OLE; NCT05648500, NCT05672576, and NCT06475742, respectively) are ongoing in patients with moderate-to-severe SLE, with the primary endpoint set as achieving an SRI-4 at 12 months. These trials evaluated the efficacy, safety, and tolerability of adding cenerimod (4 mg) to the standard treatment.
Based on previous investigations, patients with a high IFN-1 signature exhibit heightened B cell activation and T cell cooperation, indicating a greater potential significance of S1P receptor modulator regulation of lymphocyte trafficking in this population. Therefore, the precise identification of target populations, such as IFN-high or B-cell-dominant SLE subtypes, remains a challenge. From a safety perspective, concerns include not only lymphopenia but also the risks of cardiovascular events, underscoring the importance of balancing efficacy and safety compared to other agents.
FUTURE PERSPECTIVES
Unmet medical needs in SLE include maintenance of remission and achievement of cure; dependence on non-specific treatments such as GC, leading to cumulative organ damage owing to side effects; limitations in the treatment of major organ involvement such as LN and NPSLE; insufficient prognostic and disease monitoring markers; and a lack of personalized medicine.
The clinical trials conducted to date have highlighted several challenges in addressing these unmet needs. First, challenges related to clinical trial design were identified, particularly concerning the limitations of current composite endpoints, such as SRI-4 and BICLA, and their complex interactions with the heterogeneity of disease characteristics. Uniform evaluation using composite endpoints makes it challenging to assess the weighted significance of symptoms or partial improvements in diverse clinical manifestations, often emphasising changes dependent on disease activity. Many clinical trials have adopted these composite endpoints, and strict criteria may not fully reflect the actual therapeutic effects of the investigational drugs.
Moreover, there is an urgent need to establish treatment protocols that minimize GC use and include appropriate stratification based on disease activity, risk of organ damage, clear treatment goals, and the proactive introduction of immunosuppressants and biologics with demonstrated GC tapering effects. Prospective trials of GC dose reduction methods are necessary. Achieving this also depends on improving patients’ understanding of the risks associated with long-term GC use and enhancing treatment adherence.
The development of targeted therapies for refractory LN and NPSLE remains challenging. Many current clinical trials exclude patients with refractory nephritis or neuropsychiatric symptoms, emphasizing the need for clinical trials of molecular-targeted drugs, drug sensitivity testing using biological specimens, and the establishment of disease models for these challenging manifestations[72].
Future therapeutic strategies to be explored include the development of rational combination therapy regimens targeting complementary pathways, establishment of treatment approaches capable of achieving GC-free remission, and development of targeted therapies for refractory LN and NPSLE. For combination therapy regimens, the key focus will be on integrating B-cell-targeted therapies, T-cell costimulation blockers, JAK/TYK2 inhibitors, and IFN signaling inhibitors - currently being evaluated in clinical trials - into the standard treatment protocols.
Another limitation in current clinical trial designs is that patient selection is based primarily on disease activity indices, which may not effectively identify the patient subgroups that would benefit most from these novel therapies. Patient stratification is essential for the development of rational combination strategies. Some clinical trials have already begun incorporating stratification approaches. However, future studies must advance not only the assessment of drug efficacy and safety but also the identification and validation of predictive biomarkers that can help guide treatment selection and maximise therapeutic benefit[73].
SLE is characterized by high heterogeneity in both clinical manifestations and immunological abnormalities; treatment targets may differ among patients. Therefore, it is increasingly important to comprehensively consider the application of innovative clinical trial designs, such as adaptive trials that allow for flexible modifications such as dose adjustment or continuation in subgroups showing efficacy at interim analysis[74]. Other designs to be considered include basket trials that evaluate patients across different disease manifestations based on shared pathogenic pathways rather than organ-specific involvement and umbrella trials that assess the efficacy of multiple agents stratified by molecular profiles specific to particular organ involvement. The importance of actively incorporating patient-reported outcomes and quality-of-life indicators into future trials is expected to increase.
These advances require the identification of biomarkers based on a deeper understanding of disease mechanisms, which aligns with the pursuit of personalized medicine. To achieve this, progress is needed in research areas, such as single-cell and multi-omics analyses to characterise immune cell subsets, cytokine dynamics, and molecular pathways. Progress is also needed in longitudinal biomarker discovery based on treatment response, relapse, and remission data from clinical trials and real-world settings, along with genetic studies, including Genome-Wide Association Study (GWAS) and whole-genome sequencing, to identify susceptibility genes.
CONCLUSION
Recent advances in immunological research have propelled the development of novel treatments for SLE, moving closer to therapies tailored to the underlying disease mechanisms. New findings have identified potential pathogenic cells, such as Tfh cells, T peripheral helper (Tph) cells, ThA cells, Age-associated B cells, and Type 2 conventional dendritic (DC2) cells, which will likely guide the development of targeted therapies for more precise disease management. However, there remain challenges in translating these new therapeutic approaches into clinical practice, with a clear need to optimise them for diverse patient populations.
To achieve this, stratifying patients based on multiple clinical markers and developing rational methods to guide treatment are essential. Additionally, refining the disease assessment criteria from the patient's perspective could lead to more accurate and effective clinical trials, necessitating potential revisions in trial design. Ultimately, innovations in both drug development and treatment strategies, particularly those that reduce dependence on GC, hold great promise for improving SLE management.
DECLARATIONS
Authors’ contributions
Writing the manuscript: Satoh-Kanda Y
Supervised its writing: Nakayamada S, Tanaka Y
Availability of data and materials
Not applicable.
AI and AI-assisted tools Statement
We used AI-assisted language editing tools to improve clarity and grammar. The tool did not contribute to the scientific content of the study. All authors accept full responsibility for the integrity of the research and the final text.
Financial support and sponsorship
Not applicable.
Conflicts of interest
Tanaka Y has received speaking fees and/or honoraria from AbbVie, Eisai, Chugai, Eli-Lilly, Behringer-Ingelheim, GlaxoSmithKline, Taisho, AstraZeneca, Daiichi-Sankyo, Gilead, Pfizer, UCB, Asahi Kasei, Astellas, and received research grants from Behringer-Ingelheim, Taisho, and Chugai. Nakayamada S has received speaking fees and/or honoraria from GlaxoSmithKline, Astellas Pharma, Eli Lilly, AbbVie, Mitsubishi Tanabe Pharma, Janssen, Pfizer, Bristol Myers Squibb, Asahi Kasei, Chugai Pharmaceutical, AstraZeneca, Taisho Pharmaceutical, Eisai, Gilead Sciences, UCB and Ayumi and has received consulting fees, lecture fees from Asahi Kasei, Otsuka Pharmaceutical and Mitsubishi Tanabe Pharma. Tanaka Y is the Guest Editor of the Special Issue "Systemic Lupus Erythematosus: Early Diagnosis and Management Strategies" of Rare Disease and Orphan Drugs Journal. Tanaka Y was not involved in any steps of the editorial process, notably including reviewers' selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039.
3. Barber MRW, Drenkard C, Falasinnu T, et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17:515-32.
4. Arnaud L, Parodis I, Devilliers H, Chasset F. Clinical trial outcomes for SLE: what we have and what we need. Lupus Sci Med. 2024;11:e001114.
5. Franklyn K, Lau CS, Navarra SV, et al. Definition and initial validation of a Lupus Low Disease Activity State (LLDAS). Ann Rheum Dis. 2016;75:1615-21.
6. van Vollenhoven RF, Bertsias G, Doria A, et al. 2021 DORIS definition of remission in SLE: final recommendations from an international task force. Lupus Sci Med. 2021;8:e000538.
7. Fanouriakis A, Kostopoulou M, Andersen J, et al. EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann Rheum Dis. 2024;83:15-29.
8. Nakayamada S, Iwata S, Tanaka Y. Relevance of lymphocyte subsets to B cell-targeted therapy in systemic lupus erythematosus. Int J Rheum Dis. 2015;18:208-18.
9. Dörner T, Lipsky PE. The essential roles of memory B cells in the pathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2024;20:770-82.
10. Fra GP, Avanzi GC, Bartoli E. Remission of refractory lupus nephritis with a protocol including rituximab. Lupus. 2003;12:783-7.
11. Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum. 2002;46:2673-7.
12. Tokunaga M, Saito K, Kawabata D, et al. Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann Rheum Dis. 2007;66:470-5.
13. Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222-33.
14. Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64:1215-26.
15. Fanouriakis A, Kostopoulou M, Boumpas DT, et al. 2019 update of the joint european league against rheumatism and european renal association-european dialysis and transplant association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis. 2020;79:713-23.
16. Sammaritano LR, Askanase A, Mustafa RA, et al. 2024 American College of Rheumatology (ACR) guideline for the screening, treatment, and management of Lupus Nephritis. Arthritis Rheumatol. 2025;77:1115-35.
17. Iwata S, Saito K, Tokunaga M, Tanaka Y. B cell or T cell-dominant recurrence after rituximab therapy in patients with SLE. Ann Rheum Dis. 2012;71:1749-50.
18. Furie RA, Aroca G, Cascino MD, et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2022;81:100-7.
19. Chu SY, Vostiar I, Karki S, et al. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Mol Immunol. 2008;45:3926-33.
20. Singh JA, Shah NP, Mudano AS. Belimumab for systemic lupus erythematosus. Cochrane Database Syst Rev. 2021;2:CD010668.
21. Furie R, Rovin BH, Houssiau F, et al. Two-year, randomized, controlled trial of belimumab in lupus nephritis. N Engl J Med. 2020;383:1117-28.
22. Furie R, Rovin BH, Houssiau F, et al. Safety and efficacy of belimumab in patients with lupus nephritis: open-label extension of BLISS-LN study. Clin J Am Soc Nephrol. 2022;17:1620-30.
23. Rovin BH, Furie R, Teng YKO, et al. A secondary analysis of the Belimumab International Study in Lupus Nephritis trial examined effects of belimumab on kidney outcomes and preservation of kidney function in patients with lupus nephritis. Kidney Int. 2022;101:403-13.
24. Carter LM, Isenberg DA, Ehrenstein MR. Elevated serum BAFF levels are associated with rising anti-double-stranded DNA antibody levels and disease flare following B cell depletion therapy in systemic lupus erythematosus. Arthritis Rheum. 2013;65:2672-9.
25. Shipa M, Santos LR, Nguyen DX, et al. Identification of biomarkers to stratify response to B-cell-targeted therapies in systemic lupus erythematosus: an exploratory analysis of a randomised controlled trial. Lancet Rheumatol. 2023;5:e24-35.
26. Atisha-Fregoso Y, Malkiel S, Harris KM, et al. Phase II randomized trial of rituximab plus cyclophosphamide followed by belimumab for the treatment of lupus nephritis. Arthritis Rheumatol. 2021;73:121-31.
27. Agmon-Levin N, Ignatenko S, Gordienko A, et al. OP0089|phase 2 safety and efficacy of subcutaneous (S.C.) dose ianalumab (VAY736; anti-BAFFR mAb) administered every 4 weeks up to 48 weeks in patients with systemic lupus erythematosus (SLE). Ann Rheum Dis. 2024;83:140-2.
28. Dörner T, Santos Da Costa A, Oliver S, et al. POS0540 reduction of B cell subsets, autoantibodies and disease-relevant pathways in ianalumab treated patients with systemic lupus erythematosus: findings from a phase 2 clinical trial. Ann Rheum Dis. 2024;83:956-7.
29. Isenberg D, Gordon C, Licu D, Copt S, Rossi CP, Wofsy D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann Rheum Dis. 2015;74:2006-15.
30. Wu D, Li J, Xu D, et al. Telitacicept in patients with active systemic lupus erythematosus: results of a phase 2b, randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2024;83:475-87.
31. Evans LS, Lewis KE, DeMonte D, et al. Povetacicept, an enhanced dual APRIL/BAFF antagonist that modulates B lymphocytes and pathogenic autoantibodies for the treatment of lupus and other B cell-related autoimmune diseases. Arthritis Rheumatol. 2023;75:1187-202.
32. Nakayamada S, Takahashi H, Kanno Y, O'Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol. 2012;24:297-302.
33. Ramanujam M, Steffgen J, Visvanathan S, Mohan C, Fine JS, Putterman C. Phoenix from the flames: rediscovering the role of the CD40-CD40L pathway in systemic lupus erythematosus and lupus nephritis. Autoimmun Rev. 2020;19:102668.
34. Furie RA, Bruce IN, Dörner T, et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology. 2021;60:5397-407.
35. Cutcutache I, Powlesland AS, Skelton A, et al. POS0334 dapirolizumab pegol impacts important immunologic pathways in systemic lupus erythematosus: pharmacodynamic analysis of B cell and type I interferon pathways from a phase 2B trial. Ann Rheum Dis. 2024;83:265.
36. Clowse M, Isenberg D, Merrill J, et al. Dapirolizumab pegol demonstrated significant improvement in systemic lupus erythematosus disease activity: efficacy and safety results of a phase 3 trial [abstract]; 2024. Available from: https://acrabstracts.org/abstract/dapirolizumab-pegol-demonstrated-significant-improvement-in-systemic-lupus-erythematosus-disease-activity-efficacy-and-safety-results-of-a-phase-3-trial/ [Last accessed on 26 Feb 2026].
37. Karnell JL, Albulescu M, Drabic S, et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci Transl Med. 2019;11:eaar6584.
38. Furie R, Werth VP, Merola JF, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest. 2019;129:1359-71.
39. Furie RA, van Vollenhoven RF, Kalunian K, et al. Trial of anti-BDCA2 antibody litifilimab for systemic lupus erythematosus. N Engl J Med. 2022;387:894-904.
40. Werth VP, Furie RA, Romero-Diaz J, et al. Trial of anti-BDCA2 antibody litifilimab for cutaneous lupus erythematosus. N Engl J Med. 2022;387:321-31.
41. Ostendorf L, Burns M, Durek P, et al. Targeting CD38 with daratumumab in refractory systemic lupus erythematosus. N Engl J Med. 2020;383:1149-55.
42. Roccatello D, Fenoglio R, Caniggia I, et al. Daratumumab monotherapy for refractory lupus nephritis. Nat Med. 2023;29:2041-7.
43. Manzi S, Bruce IN, Morand EF, et al. Efficacy and safety of subcutaneous anifrolumab in systemic lupus erythematosus: a randomized, phase 3 study. Arthritis Rheumatol. 2025.
44. Alduraibi F, Fatima H, Hamilton JA, Chatham WW, Hsu HC, Mountz JD. Lupus nephritis correlates with B cell interferon-β, anti-Smith, and anti-DNA: a retrospective study. Arthritis Res Ther. 2022;24:87.
45. Tanaka Y, Luo Y, O'Shea JJ, Nakayamada S. Janus kinase-targeting therapies in rheumatology: a mechanisms-based approach. Nat Rev Rheumatol. 2022;18:133-45.
46. Kubo S, Yamaoka K, Kondo M, et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann Rheum Dis. 2014;73:2192-8.
47. Merrill JT, Tanaka Y, D'Cruz D, et al. Efficacy and safety of upadacitinib or elsubrutinib alone or in combination for patients with systemic lupus erythematosus: a phase 2 randomized controlled trial. Arthritis Rheumatol. 2024;76:1518-29.
48. Merrill J, Saxena A, Aringer M, et al. Efficacy and safety of ABBV-599 (elsubrutinib and upadacitinib combination) and upadacitinib monotherapy for the treatment of systemic lupus erythematosus: results through 104 weeks in a long-term extension study; 2024. Available from: https://acrabstracts.org/abstract/efficacy-and-safety-of-abbv-599-elsubrutinib-and-upadacitinib-combination-and-upadacitinib-monotherapy-for-the-treatment-of-systemic-lupus-erythematosus-results-through-104-weeks-in-a-long-term-ext/ [Last accessed on 26 Feb 2026].
49. Hasni SA, Gupta S, Davis M, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun. 2021;12:3391.
50. Dörner T, Tanaka Y, Dow ER, et al. Mechanism of action of baricitinib and identification of biomarkers and key immune pathways in patients with active systemic lupus erythematosus. Ann Rheum Dis. 2022;81:1267-72.
51. Werth VP, Fleischmann R, Robern M, et al. Filgotinib or lanraplenib in moderate to severe cutaneous lupus erythematosus: a phase 2, randomized, double-blind, placebo-controlled study. Rheumatology. 2022;61:2413-23.
52. Morand E, Merola JF, Tanaka Y, Gladman D, Fleischmann R. TYK2: an emerging therapeutic target in rheumatic disease. Nat Rev Rheumatol. 2024;20:232-40.
53. Satoh-Kanda Y, Nakayamada S, Kubo S, et al. Modifying T cell phenotypes using TYK2 inhibitor and its implications for the treatment of systemic lupus erythematosus. RMD Open. 2024;10:e003991.
54. Morand E, Pike M, Merrill JT, et al. Deucravacitinib, a tyrosine kinase 2 inhibitor, in systemic lupus erythematosus: a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2023;75:242-52.
55. Su LC, Xu WD, Huang AF. IRAK family in inflammatory autoimmune diseases. Autoimmun Rev. 2020;19:102461.
56. Isenberg D, Furie R, Jones NS, et al. Efficacy, safety, and pharmacodynamic effects of the bruton's tyrosine kinase inhibitor fenebrutinib (GDC-0853) in systemic lupus erythematosus: results of a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2021;73:1835-46.
57. Li R, Zhu X, Liu S, et al. LB0005 orelabrutinib, an irreversible inhibitor of bruton's tyrosine kinase (BTK), for the treatment of systemic lupus erythematosus (SLE): RESULTS of a randomized, double-blind, placebo-controlled, phase IB/IIA dose-finding study. Ann Rheum Dis. 2022;81:210.
58. Fillatreau S, Manfroi B, Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17:98-108.
59. Morand EF, Bender A, Deshpande A, et al. AB0444 enpatoran: preclinical evidence supporting glucocorticoid dose reduction and phase II study design in patients with SLE and/or CLE (willow). Ann Rheum Dis. 2022;81:1350-1.
60. Pearson D, Morand E, Wenzel J, et al. Randomized, placebo-controlled phase II study of enpatoran, a small molecule toll-like receptor 7/8 inhibitor, in cutaneous lupus erythematosus: results from cohort A. J Rheumatol. 2025;52 Suppl 1:1-269.
61. Morand E, Dall'Era M, Furie RA, et al. LB0004 randomised, placebo-controlled phase II study of oral enpatoran, a first-in-class toll-like receptor 7/8 inhibitor, in systemic lupus erythematosus. Ann Rheum Dis. 2025;84:316-7.
62. Tanaka Y, Kumanogoh A, Atsumi T, et al. Safety, pharmacokinetics, biomarker response and efficacy of E6742: a dual antagonist of Toll-like receptors 7 and 8, in a first in patient, randomised, double-blind, phase I/II study in systemic lupus erythematosus. RMD Open. 2024;10:e004701.
63. Rovin BH, Teng YKO, Ginzler EM, et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2021;397:2070-80.
64. Abdalhadi HM, Chatham WW, Alduraibi FK. CAR-T-cell therapy for systemic lupus erythematosus: a comprehensive overview. Int J Mol Sci. 2024;25:10511.
65. Mougiakakos D, Krönke G, Völkl S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. 2021;385:567-9.
66. Müller F, Taubmann J, Bucci L, et al. CD19 CAR T-cell therapy in autoimmune disease - a case series with follow-up. N Engl J Med. 2024;390:687-700.
67. Kollert F, Robertson S, Schuler F, et al. Characterization of RO7507062, a CD19-targeting T-cell bispecific antibody (CD19TCB), and design of its ongoing phase 1 trial in systemic lupus erythematosus; 2024. Available from: https://acrabstracts.org/abstract/characterization-of-ro7507062-a-cd19-targeting-t-cell-bispecific-antibody-cd19tcb-and-design-of-its-ongoing-phase-1-trial-in-systemic-lupus-erythematosus/ [Last accessed on 26 Feb 2026].
68. Domingo-Gonzalez R, Baribaud I, Oyasu M, et al. POS1062 therapeutic potential of imvotamab, a CD20-targeted bispecific IGM T cell engager, for the treatment of refractory autoimmune disease patients. Ann Rheum Dis. 2024;83:935.
69. Quigley M, Michaelson J, Jones J, et al. CLN-978, a CD19-directed T cell engager (TCE), leads to rapid and deep B cell depletion and has broad potential for development in autoimmune diseases; 2024. Available from: https://acrabstracts.org/abstract/cln-978-a-cd19-directed-t-cell-engager-tce-leads-to-rapid-and-deep-b-cell-depletion-and-has-broad-potential-for-development-in-autoimmune-diseases/ [Last accessed on 26 Feb 2026].
70. Alexander T, Krönke J, Cheng Q, Keller U, Krönke G. Teclistamab-induced remission in refractory systemic lupus erythematosus. N Engl J Med. 2024;391:864-6.
71. Askanase A, Berkani O, Navarra S, et al. Efficacy and safety of cenerimod in patients with moderate to severe systemic lupus erythematosus (SLE): a multicenter, randomized, parallel-group, double-blind, placebo-controlled, dose-finding phase 2b trial; 2022. Available from: https://acrabstracts.org/abstract/efficacy-and-safety-of-cenerimod-in-patients-with-moderate-to-severe-systemic-lupus-erythematosus-sle-a-multicenter-randomized-parallel-group-double-blind-placebo-controlled-dose-finding-phase/ [Last accessed on 26 Feb 2026].
72. Zhang SJ, Xu RY, Kang LL. Biomarkers for systemic lupus erythematosus: a scoping review. Immun Inflamm Dis. 2024;12:e70022.
73. Connelly K, Eades LE, Koelmeyer R, et al. Towards a novel clinical outcome assessment for systemic lupus erythematosus: first outcomes of an international taskforce. Nat Rev Rheumatol. 2023;19:592-602.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.