


A data-driven comparative study of thermomechanical properties in rare-earth zirconate and tantalate oxides for thermal barrier coatings
 0
0 Abstract
Rare-earth (RE) zirconates and tantalates are promising candidates for next-generation thermal barrier coatings (TBCs) due to their high-temperature stability and low thermal conductivity. However, the substantial compositional complexity introduced by multiple RE element substitutions poses significant challenges for systematic property optimization. To address these challenges, a high-throughput, data-driven computational framework was employed to systematically investigate and compare structural stability, thermodynamic properties, lattice thermal conductivity (κL) and fracture toughness (KIC) of RE2Zr2O7 and RE3TaO7 oxides (RE = Sc, Y, La ~ Lu) in their pyrochlore and Weberite-type structures, respectively. κL and intrinsic KIC were systematically evaluated using phonon-scattering and Griffith-based models. The results reveal that RE3TaO7 exhibits consistently lower κL than RE2Zr2O7 due to its low symmetry, heavier atomic masses and higher structural disorder. Interestingly, theoretical predictions indicate slightly higher intrinsic KIC in RE2Zr2O7, which is attributed to its ordered vacancy sublattice and symmetric bonding. In contrast, experimental data often report superior KIC for RE3TaO7, likely due to extrinsic microstructural effects not captured in idealized calculations. Correlation and SHapley Additive exPlanations analyses further reveal that bond energy, charge disorder and bond-length heterogeneity are key descriptors governing κL and KIC. These findings provide mechanistic insight into structure–property relationships and offer a predictive framework for the rational design of RE oxide TBC materials.
Highlights
• Integrating high-throughput first-principles calculations, lattice-level descriptor engineering and interpretable machine learning to design RE2Zr2O7 and RE3TaO7 (RE = Sc, Y, La ~ Lu) oxide-based thermal barrier materials.
• Data-driven selection and classification of key physical descriptors (bond energy, charge disorder, bond-length heterogeneity) enable predictive modeling of κL and KIC across 17 rare-earth elements.
• Combining thermodynamic stability analysis, phonon-based transport models and SHapley Additive exPlanations interpretability to establish structure–property relationships and guide rational oxide design.
Keywords
INTRODUCTION
Advancing the performance and reliability of thermal barrier coatings (TBCs) represents a persistent challenge in the development of next-generation high-temperature materials, particularly for demanding applications in aerospace propulsion and energy conversion systems[1-3]. The continuous push toward higher operational temperatures and harsher service environments necessitates the discovery and design of ceramic materials that not only exhibit ultralow lattice thermal conductivity (κL) for efficient thermal insulation but also possess robust mechanical stability, exemplified by high fracture toughness (KIC), along with enhanced resistance to environmental degradation[4,5]. However, conventional yttria-stabilized zirconia (YSZ), currently the industry standard, faces intrinsic limitations above 1,473 K, primarily due to phase instability and accelerated degradation mechanisms[6,7]. Consequently, exploring alternative ceramic systems with superior thermomechanical performance at elevated temperatures is of critical importance.
Rare-earth (RE)-based oxide ceramics, particularly RE zirconates and tantalates, have emerged as promising alternative candidates, owing to their intrinsically low thermal conductivity, high melting points, and exceptional structural stability at elevated temperatures[8-12]. Furthermore, the compositional flexibility afforded by substituting and doping various RE elements enables precise tuning of lattice parameters and phonon scattering characteristics, significantly optimizing their thermal and mechanical properties. Recently, high-entropy oxides (HEOs), characterized by multicomponent RE element incorporation, have attracted considerable attention due to their unique capacity for simultaneously achieving ultralow thermal conductivity and enhanced KIC, driven by severe lattice distortions and increased chemical disorder[13-15]. Nevertheless, despite these promising attributes, the complexity introduced by a large compositional space and numerous potential element combinations presents a significant obstacle to systematic optimization. This complexity necessitates an efficient and systematic approach to elucidate the influence of each RE element on thermomechanical performance and to identify optimal compositions.
Recent studies have demonstrated that κL and KIC of ceramic TBC materials are strongly influenced by atomic-scale structural characteristics, such as crystal lattice symmetry[16-18], oxygen vacancy concentration[19,20], local lattice distortion[21-24], and the nature of chemical bonding[25-27]. Reduced thermal conductivity is primarily achieved through enhanced phonon scattering mechanisms, typically arising from structural disorder, lattice anharmonicity, point defects, and atomic mass mismatch introduced via compositional complexity[21,28-30]. For example, previous investigations demonstrated that structurally complex phases, such as defect fluorite and pyrochlore structures, effectively enhance phonon scattering, thereby significantly reducing thermal conductivity[29,31]. On the other hand, KIC in ceramics predominantly depends on intrinsic factors, including bond strength, lattice rigidity, and phase stability, along with extrinsic toughening mechanisms such as crack deflection, bridging, and stress-induced phase transformations[32-34]. Controlled introduction of structural distortions and interfaces is known to activate these toughening mechanisms, thereby improving fracture resistance in brittle oxide ceramics[23,24,35]. Despite these advances, a comprehensive, systematic understanding of the individual contributions of different RE elements to the delicate balance between low κL and high KIC in zirconate and tantalate systems remains elusive. Clarifying these relationships is essential for rationally optimizing and designing advanced TBC materials with tailored thermomechanical properties.
Traditional experimental approaches to thermal barrier material discovery, typically grounded in empirical trial-and-error methodologies[36], face significant limitations when applied to the vast compositional space of RE oxides. The high cost, long cycle times and difficulty in isolating intrinsic structure–property relationships hinder the ability of these methods to identify high-performance candidates efficiently[37-39]. Moreover, most existing studies focus on individual compositions, lacking a systematic cross-comparison of the full RE series. Addressing these challenges requires an integrated strategy that can both explore broad chemical design spaces and uncover the underlying physical principles governing thermomechanical performance[40]. Advances in high-throughput computational methods combined with data-driven analyses have emerged as powerful tools for the accelerated discovery and rational design of complex, multicomponent materials[3,41,42]. By systematically computing material properties across broad chemical spaces using first-principles calculations and subsequently analyzing the resulting large datasets, these approaches enable the identification of key physical parameters and governing principles that link atomic-scale features to macroscopic behavior[43]. For instance, such approaches have identified correlations between ionic charge and thermal conductivity in perovskite oxides[44] and quantified the impact of severe lattice distortion on KIC in ceramics by linking it to local bonding environments and macroscopic elastic/plastic properties[26,27]. In addition, they have revealed how atomic bonding distortions directly influence phonon scattering and reduce thermal conductivity in RE oxides[13,27].
In this work, the present work systematically investigates and compares structural stability, κL, KIC, and bonding characteristics in RE2Zr2O7 (pyrochlore/defect fluorite) and RE3TaO7 (Weberite-type/defect fluorite) oxides across 17 RE elements (RE = Sc, Y, La ~ Lu). Although RE3TaO7 and RE2Zr2O7 oxides differ in stoichiometry and crystal symmetry, both systems feature BO6 (B = Ta or Zr) octahedra and distorted RE coordination polyhedra derived from fluorite-related frameworks. These structural analogies, together with shared controlling factors such as RE ionic radius (
MATERIALS AND METHODS
Computational workflow
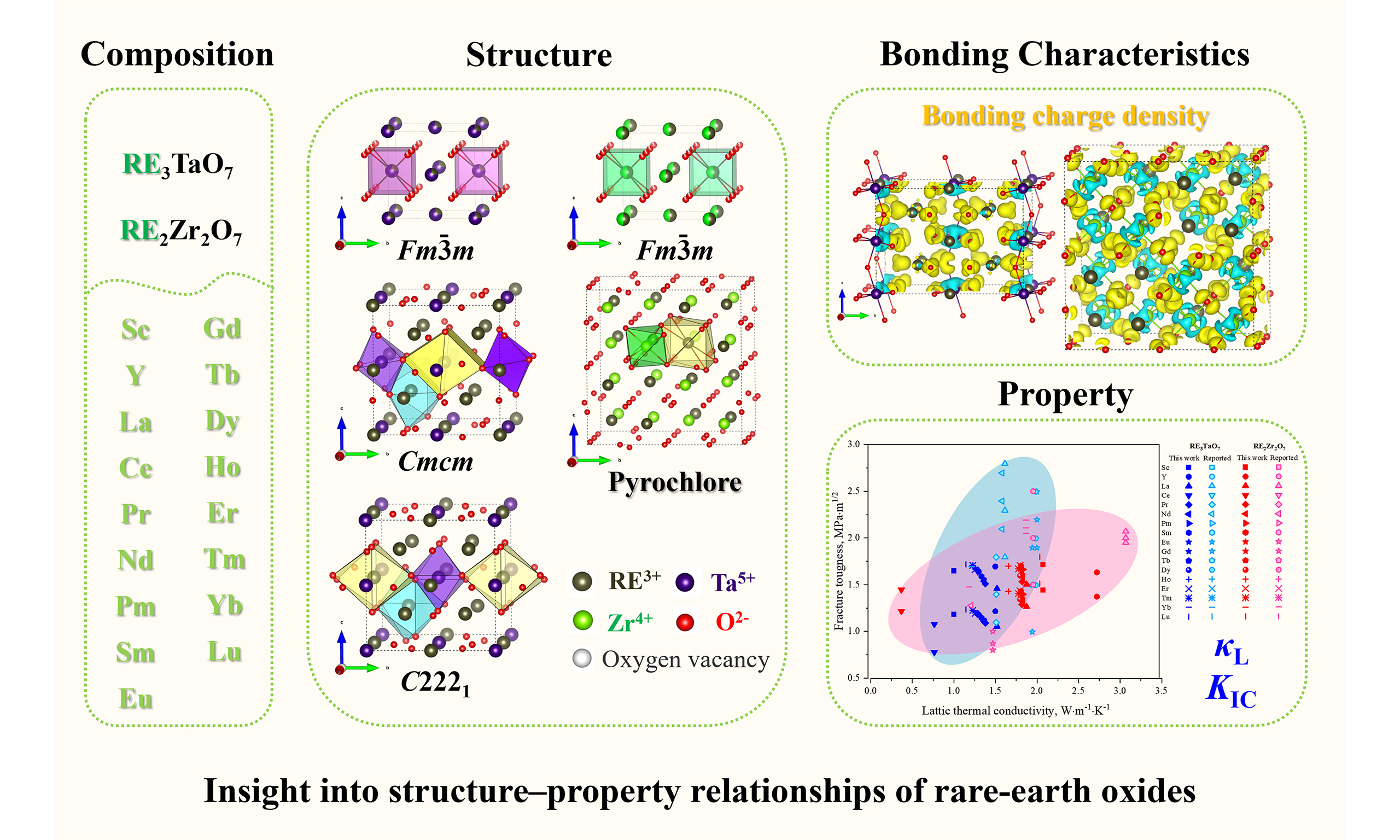
A high-throughput computational workflow was employed to establish structure–property relationships in RE3TaO7 and RE2Zr2O7 oxides, as shown in Figure 1. The workflow begins with compositional enumeration across 17 RE elements (RE = Sc, Y, La ~ Lu), followed by crystal structure selection based on predicted stable phases. Subsequently, density functional theory (DFT) calculations were performed to extract key thermodynamic and bonding descriptors. These data feed into property prediction models for KIC and κL. Finally, data-driven analyses, including principal component analysis (PCA), Pearson correlation, and SHapley Additive exPlanations (SHAP)-based interpretability methods, were applied to systematically evaluate the influence of each descriptor on the predicted KIC and κL. These analyses identify the most relevant features and provide both statistical and model-driven insight.
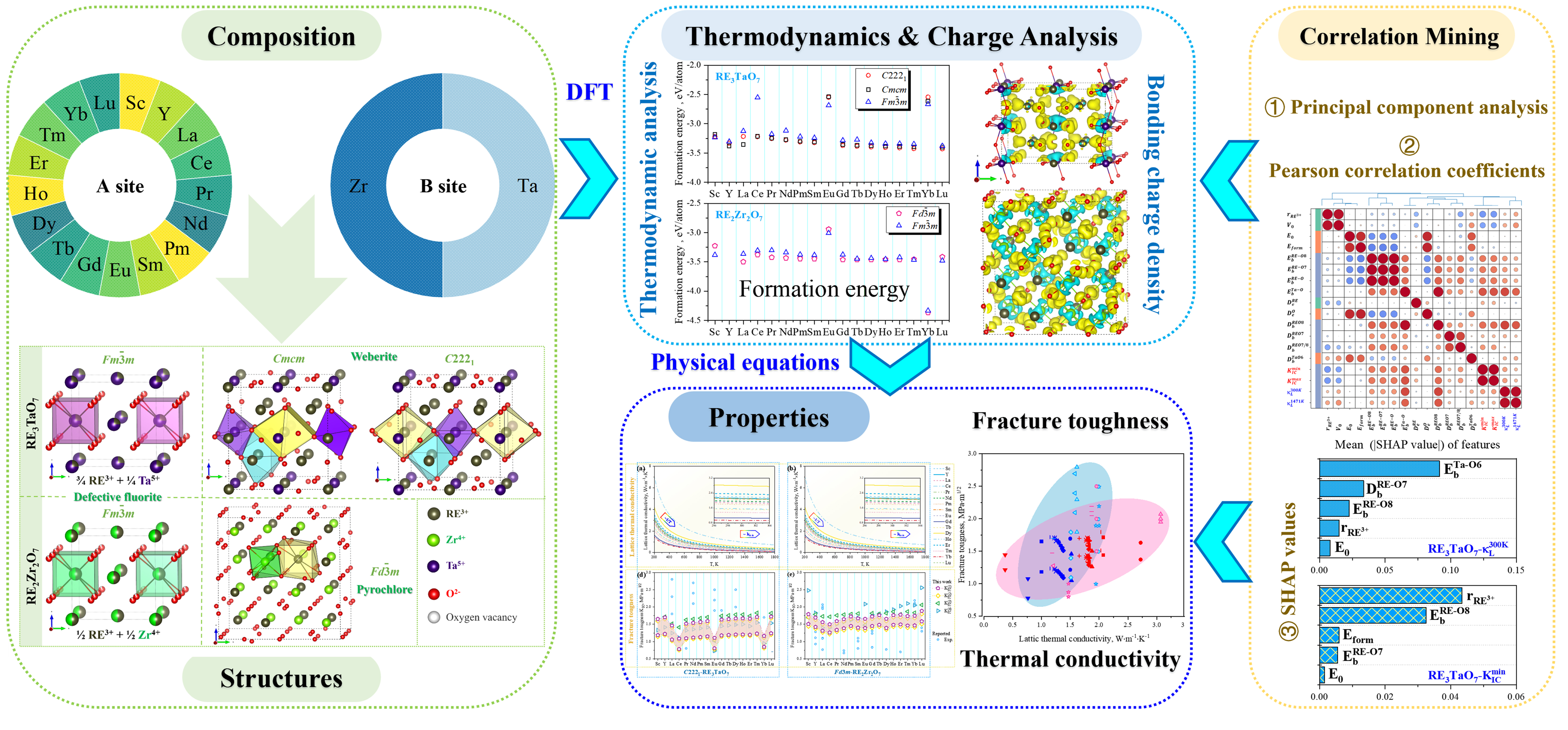
Figure 1. Schematic overview of the computational workflow for uncovering the structure–property relationships of the RE3TaO7 and RE2Zr2O7 oxides.
Crystal structure
The RE3TaO7 and RE2Zr2O7 oxide families exhibit a rich diversity of crystal structures, each governed by the ionic radius of the RE (
First-principles calculations
All DFT calculations were carried out using the projector augmented wave (PAW) method with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional within the generalized gradient approximation (GGA) as implemented in the Vienna Ab initio Simulation Package (VASP)[46]. Spin polarization was enabled, and the plane-wave energy cutoff was set to 1.4 times the maximum elemental ENMAX, where ENMAX denotes the recommended kinetic energy cutoff specified in the VASP pseudopotential files. k-point meshes of 4 × 4 × 4 ensured convergence of total energy within 10-6 eV·atom-1. Although applying Hubbard U corrections may improve prediction accuracy, the primary focus of this work is on trend-level comparisons across a wide compositional space. Several studies have established the effectiveness of GGA functionals in describing the properties of RE compounds[47-49]. Therefore, omitting U corrections introduces negligible error in the context of structural, elastic, and thermodynamic analysis. Moreover, this choice avoids the element-specific arbitrariness associated with U parameter selection and ensures methodological consistency throughout the high-throughput workflow[49]. Accordingly, all calculations in this study were carried out using the PBE functional within the GGA framework without +U corrections[50], providing a robust and computationally efficient platform for capturing structure–property relationships in RE zirconates and tantalates. The V0 and equilibrium energy (E0) were obtained by fitting the total energy–volume data to the four-parameter Birch-Murnaghan equation of state[51]. Elastic constants were obtained using the energy–strain method, from which mechanical properties including bulk modulus, shear modulus, Young’s modulus, and Poisson’s ratio were derived via the Voigt–Reuss–Hill averaging scheme[52,53].
Lattice thermal conductivity
The thermodynamic combinatorial model is employed to predict the Debye temperature Θ, Grüneisen parameters γ to the temperature-dependent κL in oxides[27]. Specifically,
have been selected as the best descriptors for calculating κL in the Slack equation:
where A is a constant, Mav is average atomic mass, n denotes the number of atoms in the unit cell, V0 represents the atomic volume. This formulation was applied to RE2Zr2O7 and RE3TaO7 oxides with temperatures ranging from 200 to 1,800 K, as reported in Ref[27].
The effective thermal conductivity κeff was normalized as
where the porosity ϕ is set to 5% in accordance with experiment[54].
Fracture toughness
The Griffith Criterion is a foundational theory in fracture mechanics for brittle materials. It postulates that crack propagation occurs when the elastic strain energy released by crack extension is sufficient to overcome the energy required to create the new crack surfaces (surface energy, γ). KIC is related to the Young’s modulus (E) and fracture energy (Gc = 2γ for mode I fracture in brittle materials) through an expression of the
where ν is Poisson’s ratio. Calculation details for the surface energy (γ) are provided in Ref[57]. It is important to note that Griffith theory generally predicts the fracture strength of materials containing pre-existing flaws, rather than the ideal theoretical strength of a perfect crystal. In practice, semi-empirical predictive models, derived from fitting experimental data, are often employed to provide correction factors for different material systems[58,59].
RESULTS AND DISCUSSION
Structural stability and thermodynamic trends
The thermodynamic stability, V0, and E0 of RE3TaO7 and RE2Zr2O7 oxides are systematically evaluated across 17 RE elements (RE = Sc, Y, La ~ Lu) using first-principles calculations in the relevant crystal structures as illustrated in Figure 2. For the RE3TaO7 oxides, V0 exhibits a consistent decreasing trend corresponding to the well-documented lanthanide contraction as the ionic radius of RE ions decreases. Among the investigated configurations, the orthorhombic C2221 phase is thermodynamically most favorable for RE3+ cations of intermediate size (e.g., Sm ~ Dy, Y), while the Cmcm structure is stabilized by the largest cations (La, Pr). The disordered fluorite structure becomes energetically competitive for smaller RE ions due to its ability to accommodate local strain through enhanced structural flexibility[60,61]. Similarly, the RE2Zr2O7 oxides also demonstrate volume contraction consistent with decreasing
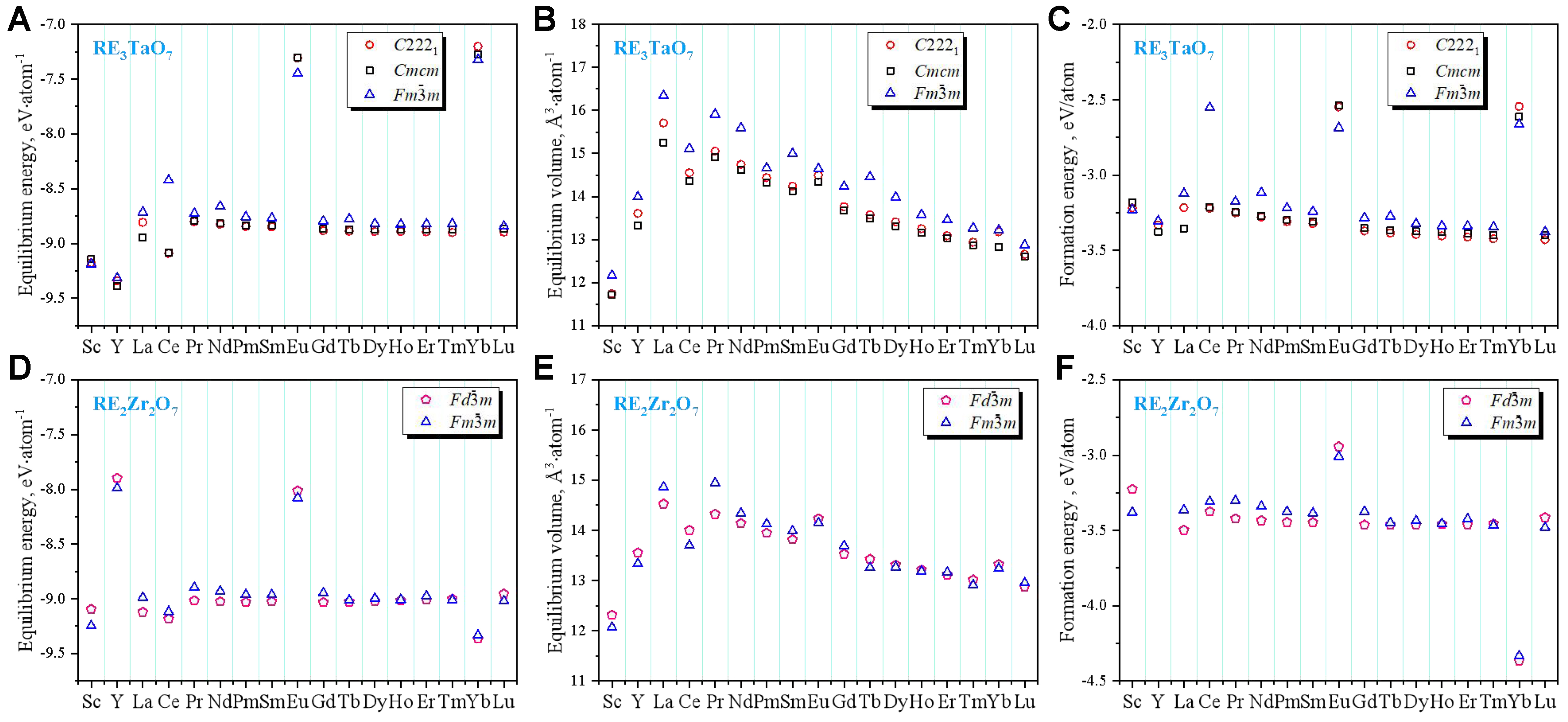
Figure 2. Thermodynamic analysis for (A-C) RE3TaO7 and (D-F) RE2Zr2O7 comparisons (RE = Sc, Y, La ~ Lu), including (A and D) Equilibrium energy, (B and E) equilibrium volume and (C and F) formation energies of different phases. Symbols denote space groups: C2221, Cmcm and Fm
To identify underlying correlations among structural and thermodynamic parameters, PCA was conducted using four standardized descriptors:
Thermal conductivity and fracture toughness
The high-temperature performance of RE2Zr2O7 and RE3TaO7 oxides as TBCs is strongly governed by their κL and KIC. In this study, κL and KIC were systematically evaluated for the pyrochlore (Fd
To ensure the validity of thermal conductivity predictions across the studied temperature range
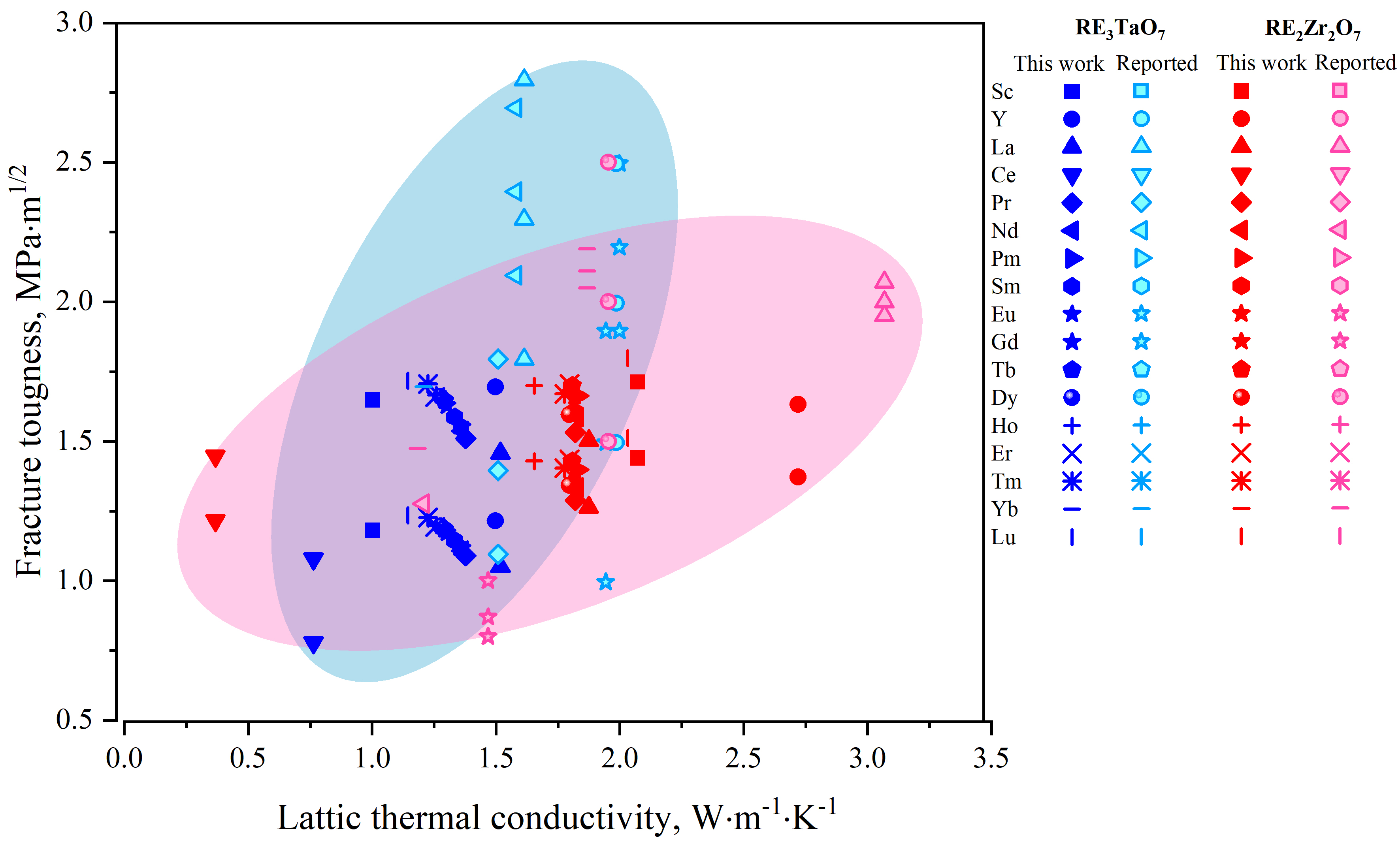
The calculated temperature-dependent κL from 200 to 1,800 K, incorporating 5% porosity corrections for representative oxides in both families is shown conceptually in Figure 3A and B. At lower temperatures, κL rapidly decreases following a typical inverse temperature dependence (1/T), governed by intrinsic phonon–phonon Umklapp scattering. At higher temperatures, κL approaches a minimum limit (κmin) due to phonon mean free paths converging toward interatomic distances, indicative of pronounced phonon scattering. As shown in Figure 3C, RE3TaO7 consistently exhibits lower κL (0.76-1.52 W·m-1·K-1 at 300 K) compared to RE2Zr2O7 (1.7-2.8 W·m-1·K-1 at 300 K), a trend maintained across the entire temperature range. The lower κL of RE3TaO7 oxides primarily results from their inherently lower symmetry (C2221), greater structural complexity and consequently increased phonon scattering. Additional factors, including larger atomic mass differences and oxygen sublattice disorder, further reduce κL by intensifying phonon scattering channels.
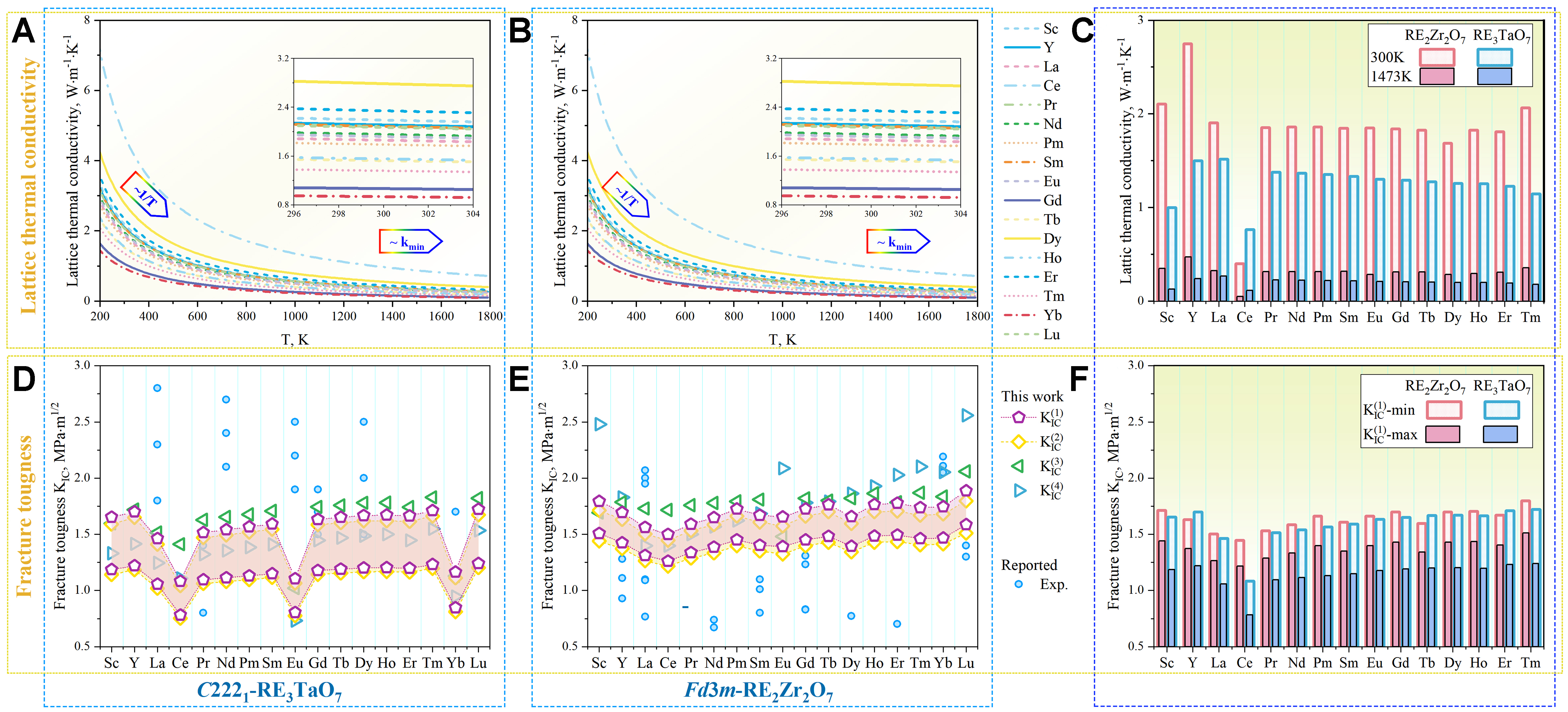
Figure 3. Temperature-dependent κL for (A) C2221 RETa3O7 and (B) RE2Zr2O7 pyrochlore oxides by the combinatorial approach (κ(2)(Θ(2)(γ(3)))) with 5% porosity correction; (C) κL values at 300 and 1,473 K. Calculated KIC by K0(n) where n = 1-4 of for (D) RE3TaO7 and (E) RE2Zr2O7 oxides, together with the reported result[69-80]; (F) Comparison of the minimum and maximum predicted KIC values by K0(1).
For the identification of the optimal model for predicting KIC, Figure 3D-F compares the results obtained from various models (K0(1) and K0(2) for energetic, K0(3) and K0(4) for empirical) for oxides, alongside the reported results. Among these models, K0(1) =
Figure 3F summarizes these trends, highlighting that while RE2Zr2O7 oxides demonstrate uniformly high intrinsic KIC across the RE series, the C2221-type RE3TaO7 oxides exhibit greater variability, with maximum toughness predicted for Tm3TaO7 and Lu3TaO7. The superior intrinsic performance of RE2Zr2O7 is closely linked to its symmetric bonding network and ordered defect structure, which provide effective energy dissipation near crack tips[64]. In contrast, the more complex and disordered lattice of RE3TaO7 limits such toughening routes under idealized conditions. Moreover, the combined plot of KIC vs. κL at room temperature directly illustrates the inherent trade-off between mechanical robustness and thermal insulation, as shown in Figure 4. RE2Zr2O7 oxides cluster within a region of high KIC and moderate thermal conductivity, ideal for applications prioritizing mechanical integrity. In contrast, the orthorhombic C2221-type RE3TaO7 oxides occupy the low-κL, moderate-KIC region, which is advantageous for maximizing thermal insulation, albeit potentially at the expense of mechanical robustness. These results, validated against available experimental data, underscore the effectiveness of the data-driven predictive framework employed.
Atomic and electronic attributes of lattice distortion
To clarify the atomistic origins governing the thermal conductivity and KIC of RE-based RE3TaO7 and RE2Zr2O7 oxides, detailed analyses of bonding environments were conducted. These include bonding charge density distributions and quantitative descriptors related to bond length and charge heterogeneity. Such atomic-scale characterizations directly inform the macroscopic property trends observed, offering a unified descriptor-based framework justified by the structural similarities between the two oxide families - both composed of interconnected BO6 octahedra (B = Ta or Zr) and distorted RE–O polyhedra, despite differences in nominal RE:O ratios.
As shown in Figure 5, bonding charge density (Δρ)[65] isosurfaces reveal marked differences in electronic distribution between the orthorhombic C2221-type RE3TaO7 and pyrochlore RE2Zr2O7 oxides. In both cases, yellow and blue regions around RE and O atoms indicate strong ionic character. However, the RE3TaO7 oxides exhibit pronounced anisotropy in charge distribution, consistent with their lower crystal symmetry and indicative of stronger local lattice distortions. In contrast, the zirconates show more isotropic and symmetric Δρ patterns, aligned with their higher lattice symmetry and more ordered vacancy configuration.
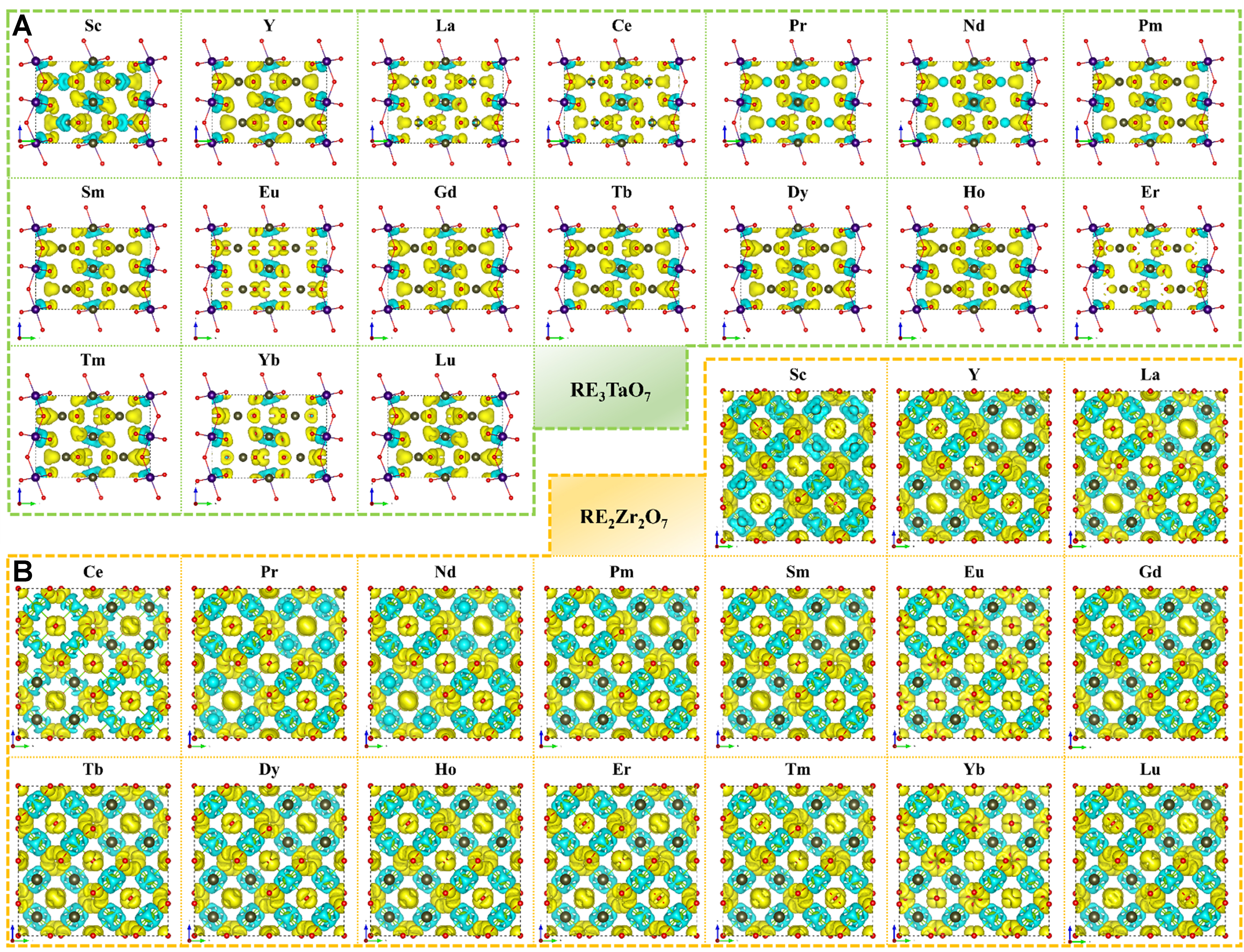
Figure 5. The bonding charge density isosurface of (A) C2221 RETa3O7 and (B) RE2Zr2O7 pyrochlore (RE = Sc, Y, La ~ Lu). The blue and yellow isosurfaces respectively correspond to Δρ ± 0.02 e-Å-3. RE: Rare-earth.
To quantitatively assess local structural disorder, two descriptors were defined: bond-length variance (Db) and Bader charge variance (De):
where bi and ei represent individual bond lengths and Bader charges within RE–O or Zr/Ta–O coordination environments, and
As shown in Figure 6A, the relative bond lengths across different RE–O and Zr/Ta–O coordination units (REO7, REO8, and mixed REO7/8 polyhedra) decrease with decreasing
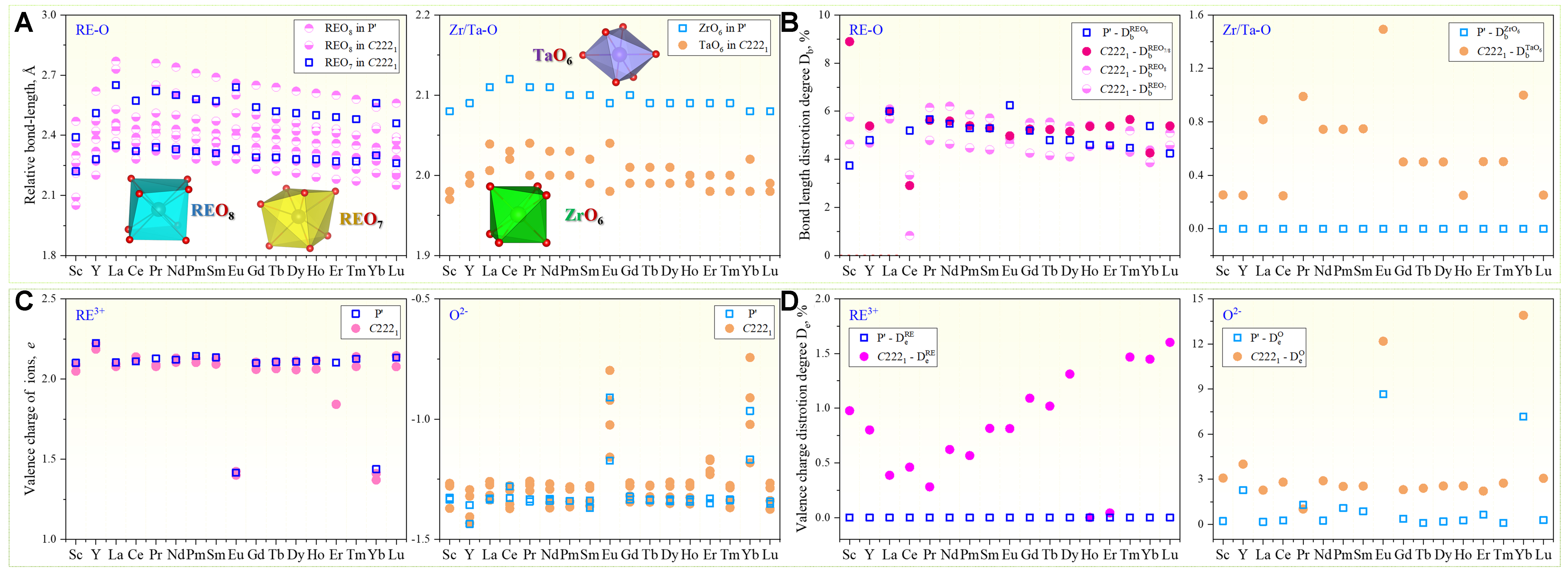
Figure 6. Bonding and charge disorder analysis in RE3TaO7 and RE2Zr2O7 oxides. (A) Relative bond lengths for RE–O (left) and Zr/Ta–O (right) polyhedra, where “REO7”, “REO8” and “REO7/8” denote the 7- and 8-fold and mixed-coordination RE–O environments. Insets illustrate representative polyhedral units; (B) Bond-length disorder degree Db of RE–O (left) and Zr/Ta–O (right) polyhedra, using the same symbol scheme as in (A); (C) Valence charges of cations RE3+ (left) and anions O2- (right), derived from Bader charge analysis; (D) Valence charge disorder degree De for RE3+ (DeRE) and O2- (DeO), quantifying charge inhomogeneity within coordination polyhedra. RE: Rare-earth.
In Figure 6C and D, electronic structure insights from Bader charge analysis further clarify structure–property relationships. Most RE ions exhibit Bader charges consistent with a nominal +3 oxidation state. However, Eu and Yb in the RE3TaO7 system show significantly lower calculated charges, indicative of a preference for the divalent (+2) state. As shown in Figure 6D, this deviation introduces local charge imbalance and strain fields, broadening the bond-length distribution and increasing De, which in turn destabilizes the ordered C2221 structure (reflected in higher relative Eform, as shown in Figure 2). Interestingly, such pronounced effects are not observed in the corresponding zirconates, suggesting the more flexible fluorite-derived framework of the zirconates is better able to accommodate such charge and strain fluctuations. These mechanisms provide vital guidelines for strategically balancing KIC and thermal insulation in RE-based oxide ceramics for advanced TBC applications.
Quantifying structure–property relationships
To establish direct links between atomic-scale descriptors and macroscopic thermomechanical behavior, quantitative correlation and interpretability analyses were conducted. Figure 7 presents Pearson correlation coefficient matrices and SHAP feature importance rankings for both RE3TaO7 and RE2Zr2O7 oxides. The SHAP package was utilized to interpret the ML models effectively[68]. These tools collectively identify the most influential parameters controlling κL and KIC, including the
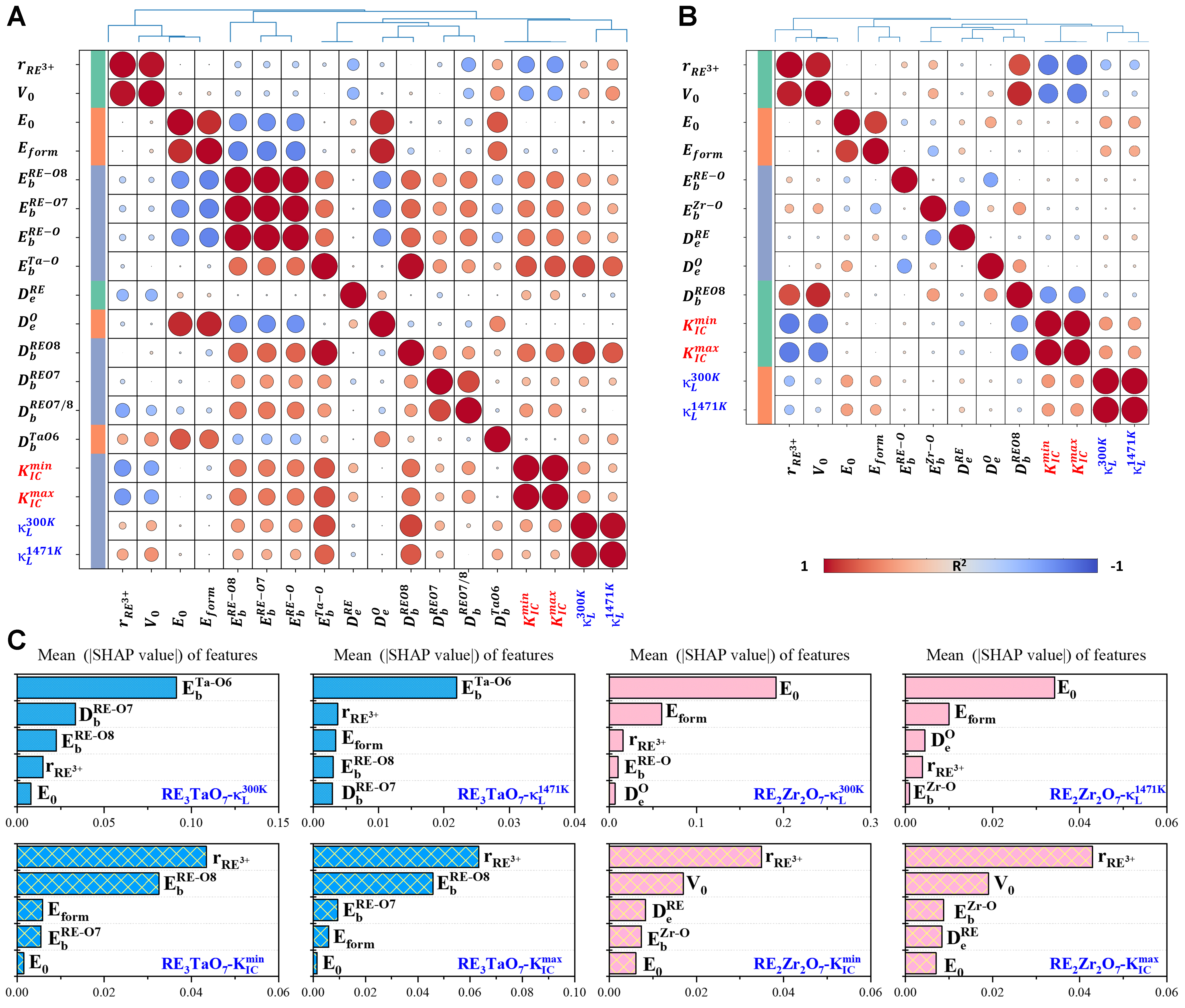
Figure 7. Correlation analysis across RE3TaO7 and RE2Zr2O7 between target properties and fundamental descriptors, including are-earth ionic radius (
In RE3TaO7 oxides, as shown in Figure 7A, strong positive correlations are observed between
To further quantify the relative importance of these descriptors, SHAP analysis was applied to trained regression models for each oxide family, as shown in Figure 7C. For RE3TaO7, dominant predictive features include RE–O Eb, bond-length disorder (Db), and Eform - consistent with the physical picture of configurational complexity and bond robustness as key regulators of performance. In RE2Zr2O7, the most influential features shift slightly to Zr–O Eb, Bader charge disorder, and V0, emphasizing the role of atomic packing and local charge environment. Taken together, both correlation-based and model-based analyses converge on three dominant descriptors: Eb, Db and Bader charge heterogeneity (De), as the primary determinants of KIC and κL across the RE series. These interpretable, physically grounded parameters elucidate the mechanisms of phonon scattering, lattice stiffening and crack resistance. Moreover, they serve as a robust foundation for data-driven materials design. Importantly, these insights explain why RE3TaO7 oxides, despite exhibiting intrinsically lower thermal conductivity, maintain moderate toughness. The reinforcement of RE–O bonds and local disorder-induced energy dissipation mechanisms helps compensate for structural complexity. In contrast, RE2Zr2O7 oxides achieve a superior balance of high fracture resistance and moderate κL, attributable to their chemically ordered and structurally accommodating lattice framework.
CONCLUSIONS
In this work, a comprehensive data-driven framework was employed to investigate the thermomechanical performance of RE3TaO7 and RE2Zr2O7 oxides, aiming to guide the rational design of advanced TBC materials. First-principles calculations and model-based analyses were used to evaluate their structural stability, thermal conductivity, and intrinsic KIC across the RE series. The results reveal that RE3TaO7 exhibits consistently lower κL than RE2Zr2O7, primarily due to its lower crystallographic symmetry, heavier atomic constituents, and higher degree of structural disorder. In contrast, RE2Zr2O7 displays slightly higher intrinsic KIC, which is attributed to its ordered vacancy sublattice and more symmetric bonding environment. However, this theoretical trend differs from experimental observations, where RE3TaO7 ceramics often demonstrate comparable or higher KIC, likely due to extrinsic microstructural effects such as porosity, residual stress, and grain boundary interactions, which are not captured in idealized simulations. To uncover the underlying structure–property relationships, both Pearson correlation and SHAP analyses were conducted, highlighting Eb, charge disorder and bond-length heterogeneity as the most influential descriptors governing κL and KIC. These insights not only reconcile theoretical predictions with experimental data but also provide a predictive framework for identifying and optimizing RE oxide compositions with balanced mechanical integrity and thermal insulation performance for high-temperature applications.
DECLARATIONS
Acknowledgments
This work was financially supported by the National Defense Basic Scientific Research Program (Grant Nos. 211-CXCY-N103-03-04-00 and 2022-JCKY-JJ-1086) and the National Natural Science Foundation of China (Grant No. 52204343). First-principles calculations were performed on the clusters at Northwestern Polytechnical University.
Authors’ contributions
Writing - original draft preparation, conceptualization, methodology, data curation, investigation, formal analysis: Zhang, Y.
Writing - original draft preparation, supervision, methodology, editing, validation, project administration, funding acquisition: Wang, W. Y.
Data curation, investigation, formal analysis, writing - original draft preparation: Wang, Y.; Ren, K.; Wang, Z.
Conceptualization, methodology, editing, project administration: Zhang, K.
Supervision, conceptualization, methodology, editing, project administration: Gao, X.; Song, H.
Supervision, conceptualization, methodology, editing, project administration, funding acquisition: Liang, X.; Li, J.
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Financial support and sponsorship
This work was financially supported by the National Defense Basic Scientific Research Program (Grant Nos. 211-CXCY-N103-03-04-00 and 2022-JCKY-JJ-1086).
Conflicts of interest
Wang, W. Y. is an Editor in the Junior Editorial Board of Journal of Materials Informatics. Wang, W. Y. was not involved in any steps of the editorial process, notably including the selection of reviewers, manuscript handling, or decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Clarke, D. R. Materials selection guidelines for low thermal conductivity thermal barrier coatings. Surf. Coat. Technol. 2003, 163-4, 67-74.
2. Padture, N. P.; Gell, M.; Jordan, E. H. Thermal barrier coatings for gas-turbine engine applications. Science 2002, 296, 280-4.
3. Ashofteh, A.; Rajabzadeh, M. Advances in thermal barrier coatings modeling, simulation, and analysis: a review. J. Eur. Ceram. Soc. 2024, 44, 116693.
4. Kumar, V.; Balasubramanian, K. Progress update on failure mechanisms of advanced thermal barrier coatings: a review. Prog. Org. Coat. 2016, 90, 54-82.
6. Song, D.; Ryu, M.; Kwon, J.; et al. Blocking of radiative thermal conduction in Zn2+-incorporated high-entropy A2B2O7 fluorite oxides. Ceram. Int. 2021, 47, 33544-53.
7. Schelling, P. K.; Phillpot, S. R.; Wolf, D. Mechanism of the cubic‐to‐tetragonal phase transition in zirconia and yttria‐stabilized zirconia by molecular‐dynamics simulation. J. Am. Ceram. Soc. 2001, 84, 1609-19.
8. Xiang, H.; Xing, Y.; Dai, F.; et al. High-entropy ceramics: present status, challenges, and a look forward. J. Adv. Ceram. 2021, 10, 385-441.
9. Wu, S.; Zhao, Y.; Li, W.; Liu, W.; Wu, Y.; Liu, F. Research progresses on ceramic materials of thermal barrier coatings on gas turbine. Coatings 2021, 11, 79.
10. Chen, L.; Hu, M.; Wang, J.; Li, B.; Feng, J. Dominant mechanisms of thermo-mechanical properties of weberite-type RE3TaO7 (RE = La, Pr, Nd, Eu, Gd, Dy) tantalates toward multifunctional thermal/environmental barrier coating applications. Acta. Mater. 2024, 270, 119857.
11. Chen, L.; Li, B.; Feng, J. Rare-earth tantalates for next-generation thermal barrier coatings. Prog. Mater. Sci. 2024, 144, 101265.
12. Haoming, Z.; Hongsong, Z.; Xiaoqin, G.; et al. Phase compositions and thermophysical performances for (Sm1-xYbx)3TaO7 compounds. Cerams. Int. 2024, 50, 18576-83.
13. Riffe, W. T.; Zare, S.; Ardrey, K. D.; et al. Broadband optical phonon scattering reduces the thermal conductivity of multi-cation oxides. Nat. Commun. 2025, 16, 3333.
14. Jia, H.; Li, C.; Chen, G.; Gong, B.; An, L.; Chen, K. Thermodynamic calculation, preparation and properties of Y2(Zr1/6Ti1/3Ge1/6Hf1/12Sn1/4)2O7 high-entropy pyrochlore ceramics. Ceram. Int. 2024, 50, 22671-8.
15. Wei, M.; Xu, J.; Zhu, J.; et al. Influence of size disorder parameter on the thermophysical properties of rare‐earth‐zirconate medium‐entropy ceramics. J. Am. Ceram. Soc. 2023, 106, 2037-48.
16. Wan, C.; Qu, Z.; Du, A.; Pan, W. Order–disorder transition and unconventional thermal conductivities of the (Sm1-xYbx)2Zr2O7 series. J. Am. Ceram. Soc. 2011, 94, 592-6.
17. Ren, S.; Zong, H. X.; Tao, X. F.; et al. Boson-peak-like anomaly caused by transverse phonon softening in strain glass. Nat. Commun. 2021, 12, 5755.
18. Wright, A. J.; Wang, Q.; Hu, C.; Yeh, Y.; Chen, R.; Luo, J. Single-phase duodenary high-entropy fluorite/pyrochlore oxides with an order-disorder transition. Acta. Mater. 2021, 211, 116858.
19. Teng, Z.; Tan, Y.; Zeng, S.; et al. Preparation and phase evolution of high-entropy oxides A2B2O7 with multiple elements at A and B sites. J. Eur. Ceram. Soc. 2021, 41, 3614-20.
20. Ren, G.; Zhang, H.; Che, J.; et al. Oxygen ion diffusion in RE3TaO7: why long-range migration of O2- is prohibited in the defective-fluorite structure? Acta. Mater. 2024, 281, 120362.
21. Wright, A. J.; Wang, Q.; Ko, S.; Chung, K. M.; Chen, R.; Luo, J. Size disorder as a descriptor for predicting reduced thermal conductivity in medium- and high-entropy pyrochlore oxides. Scr. Mater. 2020, 181, 76-81.
22. Toher, C.; Oses, C.; Esters, M.; et al. High-entropy ceramics: propelling applications through disorder. MRS. Bull. 2022, 47, 194-202.
23. Han, Y.; Liu, X.; Zhang, Q.; et al. Ultra-dense dislocations stabilized in high entropy oxide ceramics. Nat. Commun. 2022, 13, 2871.
24. Yang, Y.; Song, Z.; Lu, G.; et al. Intrinsic toughening and stable crack propagation in hexagonal boron nitride. Nature 2021, 594, 57-61.
25. Lee, S.; Esfarjani, K.; Luo, T.; Zhou, J.; Tian, Z.; Chen, G. Resonant bonding leads to low lattice thermal conductivity. Nat. Commun. 2014, 5, 3525.
26. Singh, P.; Vela, B.; Ouyang, G.; et al. A ductility metric for refractory-based multi-principal-element alloys. Acta. Mater. 2023, 257, 119104.
27. Zhang, Y.; Ren, K.; Wang, W. Y.; et al. Discovering the ultralow thermal conductive A2B2O7-type high-entropy oxides through the hybrid knowledge-assisted data-driven machine learning. J. Mater. Sci. Technol. 2024, 168, 131-42.
28. Gu, H.; Rohmer, J.; Jetter, J.; et al. Exploding and weeping ceramics. Nature 2021, 599, 416-20.
29. Braun, J. L.; Rost, C. M.; Lim, M.; et al. Charge-induced disorder controls the thermal conductivity of entropy-stabilized oxides. Adv. Mater. 2018, 30, e1805004.
30. Sarkar, A.; Wang, Q.; Schiele, A.; et al. High-entropy oxides: fundamental aspects and electrochemical properties. Adv. Mater. 2019, 31, e1806236.
31. He, J.; Xia, Y.; Lin, W.; et al. Accelerated discovery and design of ultralow lattice thermal conductivity materials using chemical bonding principles. Adv. Funct. Mater. 2022, 32, 2108532.
33. Sun, C.; Huang, Y.; Shen, Q.; et al. Embedding two-dimensional graphene array in ceramic matrix. Sci. Adv. 2020, 6, eabb1338.
34. Ritchie, R. O. Toughening materials: enhancing resistance to fracture. Philos. Trans. A. Math. Phys. Eng. Sci. 2021, 379, 20200437.
35. Porz, L.; Klomp, A. J.; Fang, X.; et al. Dislocation-toughened ceramics. Mater. Horiz. 2021, 8, 1528-37.
36. Han, J.; Kim, I.; Cho, N.; et al. Toward accurate machine learning-driven prediction of polymeric composites properties based on experimental data. MGE. Adv. 2025, 3, e70027.
37. Wang, W. Y.; Zhang, S.; Li, G.; et al. Artificial intelligence enabled smart design and manufacturing of advanced materials: the endless frontier in AI+ era. MGE. Adv. 2024, 2, e56.
38. Shang, Y.; Xiong, Z.; An, K.; Hauch, J. A.; Brabec, C. J.; Li, N. Materials genome engineering accelerates the research and development of organic and perovskite photovoltaics. MGE. Adv. 2024, 2, e28.
39. Wang, W. Y.; Yin, J.; Chai, Z.; et al. Big data-assisted digital twins for the smart design and manufacturing of advanced materials: from atoms to products. J. Mater. Inf. 2022, 2, 1.
40. Gao, X.; Wang, W. Y.; Chen, X.; et al. ProME: an integrated computational platform for material properties at extremes and its application in multicomponent alloy design. MGE. Adv. 2025, 3, e70029.
41. Divilov, S.; Eckert, H.; Hicks, D.; et al. Disordered enthalpy-entropy descriptor for high-entropy ceramics discovery. Nature 2024, 625, 66-73.
42. Xu, D.; Zhang, Q.; Huo, X.; Wang, Y.; Yang, M. Advances in data-assisted high-throughput computations for material design. MGE. Adv. 2023, 1, e11.
43. Zhang, S.; Wang, W. Y.; Wang, X.; et al. Large language models enabled intelligent microstructure optimization and defects classification of welded titanium alloys. J. Mater. Inf. 2024, 4, 34.
44. Carrete, J.; Li, W.; Mingo, N.; Wang, S.; Curtarolo, S. Finding unprecedentedly low-thermal-conductivity half-heusler semiconductors via high-throughput materials modeling. Phys. Rev. X. 2014, 4, 011019.
45. Li, Y.; Kowalski, P. M.; Beridze, G.; Birnie, A. R.; Finkeldei, S.; Bosbach, D. Defect formation energies in A2B2O7 pyrochlores. Scr. Mater. 2015, 107, 18-21.
46. Wang, Y.; Perdew, J. P. Correlation hole of the spin-polarized electron gas, with exact small-wave-vector and high-density scaling. Phys. Rev. B. Condens. Matter. 1991, 44, 13298-307.
47. Amari, S.; Daoud, S. Structural phase transition, elastic constants and thermodynamic properties of TmAs: a DFT study. Comput. Condens. Matter. 2022, 33, e00764.
48. Söderlind, P.; Turchi, P. E.; Landa, A.; Lordi, V. Ground-state properties of rare-earth metals: an evaluation of density-functional theory. J. Phys. Condens. Matter. 2014, 26, 416001.
49. Loschen, C.; Carrasco, J.; Neyman, K. M.; Illas, F. First-principles LDA+U and GGA+U study of cerium oxides: dependence on the effective U parameter. Phys. Rev. B. 2007, 75, 035115.
50. Singh, P.; Del Rose, T.; Vazquez, G.; Arroyave, R.; Mudryk, Y. Machine-learning enabled thermodynamic model for the design of new rare-earth compounds. Acta. Mater. 2022, 229, 117759.
52. Chung, D. H.; Buessem, W. R. The Voigt-Reuss-Hill approximation and elastic moduli of polycrystalline MgO, CaF2, β-ZnS, ZnSe, and CdTe. J. Appl. Phys. 1967, 38, 2535-40.
53. Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A. 1952, 65, 349-54.
54. Wan, C.; Zhang, W.; Wang, Y.; et al. Glass-like thermal conductivity in ytterbium-doped lanthanum zirconate pyrochlore. Acta. Mater. 2010, 58, 6166-72.
55. To, T.; Sørensen, S. S.; Stepniewska, M.; et al. Fracture toughness of a metal-organic framework glass. Nat. Commun. 2020, 11, 2593.
56. Griffith, A. A. VI. The phenomena of rupture and flow in solids. Philos. Trans. A. Math. Phys. Eng. Sci. 1921, 221, 163-98.
57. Zhang, Y.; Wang, W. Y.; Li, P.; et al. Hook’s law scaled broken-bond model for surface energy: from metals to ceramics. Scr. Mater. 2024, 244, 116026.
58. Niu, H.; Niu, S.; Oganov, A. R. Simple and accurate model of fracture toughness of solids. J. Appl. Phys. 2019, 125, 065105.
59. Mazhnik, E.; Oganov, A. R. A model of hardness and fracture toughness of solids. J. Appl. Phys. 2019, 126, 125109.
60. Wang, J.; Zhang, F.; Lian, J.; Ewing, R. C.; Becker, U. Energetics and concentration of defects in Gd2Ti2O7 and Gd2Zr2O7 pyrochlore at high pressure. Acta. Mater. 2011, 59, 1607-18.
61. Shamblin, J.; Tracy, C. L.; Palomares, R. I.; et al. Similar local order in disordered fluorite and aperiodic pyrochlore structures. Acta. Mater. 2018, 144, 60-7.
62. Zhang, Y.; Ren, K.; Wang, W. Y.; et al. Smart design A2Zr2O7-type high-entropy oxides through lattice-engineering toughening strategy. npj. Comput. Mater. 2024, 10, 1462.
63. Yokogawa, Y.; Yoshimura, M. Formation and stability regions of the high‐temperature fluorite‐related phase in the R2O3‐Ta2O5 system (R = La, Nd, Sm, Ho, Er, and Yb). J. Am. Ceram. Soc. 1997, 80, 1965-74.
64. Labrincha, J. A.; Frade, J. R.; Marques, F. M. B. La2Zr2O7 formed at ceramic electrode/YSZ contacts. J. Mater. Sci. 1993, 28, 3809-15.
65. Su, L.; Huyan, H.; Sarkar, A.; et al. Direct observation of elemental fluctuation and oxygen octahedral distortion-dependent charge distribution in high entropy oxides. Nat. Commun. 2022, 13, 2358.
66. Cui, K.; Sun, T. L.; Liang, X.; et al. Multiscale energy dissipation mechanism in tough and self-healing hydrogels. Phys. Rev. Lett. 2018, 121, 185501.
67. Cui, K.; Ye, Y. N.; Sun, T. L.; et al. Effect of structure heterogeneity on mechanical performance of physical polyampholytes hydrogels. Macromolecules 2019, 52, 7369-78.
69. Zhao, X.; Guo, L.; Wang, C.; Zhang, Y.; Ye, F. Effect of phase structure evolution on thermal expansion and toughness of (Nd1-xScx)2Zr2O7 (x = 0, 0.025, 0.05, 0.075, 0.1) ceramics. J. Mater. Sci. Technol. 2017, 33, 192-7.
70. Hua, Y.; Jiang, B.; Chen, R.; Cao, J.; Shuai, W.; Li, R. Enhanced physical properties of TiSi2 doped Gd2Zr2O7 ceramic for thermal barrier coatings. Mater. Res. Express. 2019, 6, 056547.
71. Wang, C.; Guo, L.; Zhang, Y.; Zhao, X.; Ye, F. Enhanced thermal expansion and fracture toughness of Sc2O3-doped Gd2Zr2O7 ceramics. Ceram. Int. 2015, 41, 10730-5.
72. Tu, T.; Liu, J.; Zhou, L.; Liang, Y.; Zhang, G. Graceful behavior during CMAS corrosion of a high-entropy rare-earth zirconate for thermal barrier coating material. J. Eur. Ceram. Soc. 2022, 42, 649-57.
73. Liu, D.; Shi, B.; Geng, L.; Wang, Y.; Xu, B.; Chen, Y. High-entropy rare-earth zirconate ceramics with low thermal conductivity for advanced thermal-barrier coatings. J. Adv. Ceram. 2022, 11, 961-73.
74. Mao, W.; Wang, Y.; Huang, H.; et al. In situ characterizations of mechanical behaviors of freestanding (Gd0.9Yb0.1)2Zr2O7 coatings by bending tests under different temperatures based on digital image correlation. J. Eur. Ceram. Soc. 2020, 40, 491-502.
75. Wang, D.; Dong, S.; Zeng, J.; et al. Influence of doping Mg2+ or Ti4+ captions on the microstructures, thermal radiation and thermal cycling behavior of plasma-sprayed Gd2Zr2O7 coatings. Ceram. Int. 2020, 46, 13054-65.
76. Ren, X.; Wan, C.; Zhao, M.; Yang, J.; Pan, W. Mechanical and thermal properties of fine-grained quasi-eutectoid (La1-xYbx)2Zr2O7 ceramics. J. Eur. Ceram. Soc. 2015, 35, 3145-54.
77. Yan, R.; Liang, W.; Miao, Q.; et al. Mechanical, thermal and CMAS resistance properties of high-entropy (Gd0.2Y0.2Er0.2Tm0.2Yb0.2)2Zr2O7 ceramics. Ceram. Int. 2023, 49, 20729-41.
78. Ren, K.; Wang, Q.; Shao, G.; Zhao, X.; Wang, Y. Multicomponent high-entropy zirconates with comprehensive properties for advanced thermal barrier coating. Scr. Mater. 2020, 178, 382-6.
79. Guo, L.; Zhang, Y.; Zhao, X.; Wang, C.; Ye, F. Thermal expansion and fracture toughness of (RE0.9Sc0.1)2Zr2O7 (RE = La, Sm, Dy, Er) ceramics. Ceram. Int. 2016, 42, 583-8.
80. Zhang, Y.; Guo, L.; Zhao, X.; Wang, C.; Ye, F. Toughening effect of Yb2O3 stabilized ZrO2 doped in Gd2Zr2O7 ceramic for thermal barrier coatings. Mater. Sci. Eng. A. 2015, 648, 385-91.
81. Guo, L.; Guo, H.; Peng, H.; Gong, S. Thermophysical properties of Yb2O3 doped Gd2Zr2O7 and thermal cycling durability of (Gd0.9Yb0.1)2Zr2O7/YSZ thermal barrier coatings. J. Eur. Ceram. Soc. 2014, 34, 1255-63.
82. Wu, Y.; Zheng, L.; He, W.; He, J.; Guo, H. Effects of Yb3+ doping on phase structure, thermal conductivity and fracture toughness of (Nd1-xYbx)2Zr2O7. Ceram. Int. 2019, 45, 3133-9.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.