

Rewiring brain iron biology: development, plasticity and disease
Keywords: iron metabolism, brain iron, ferroplasticity, neural development, therapeutic targeting
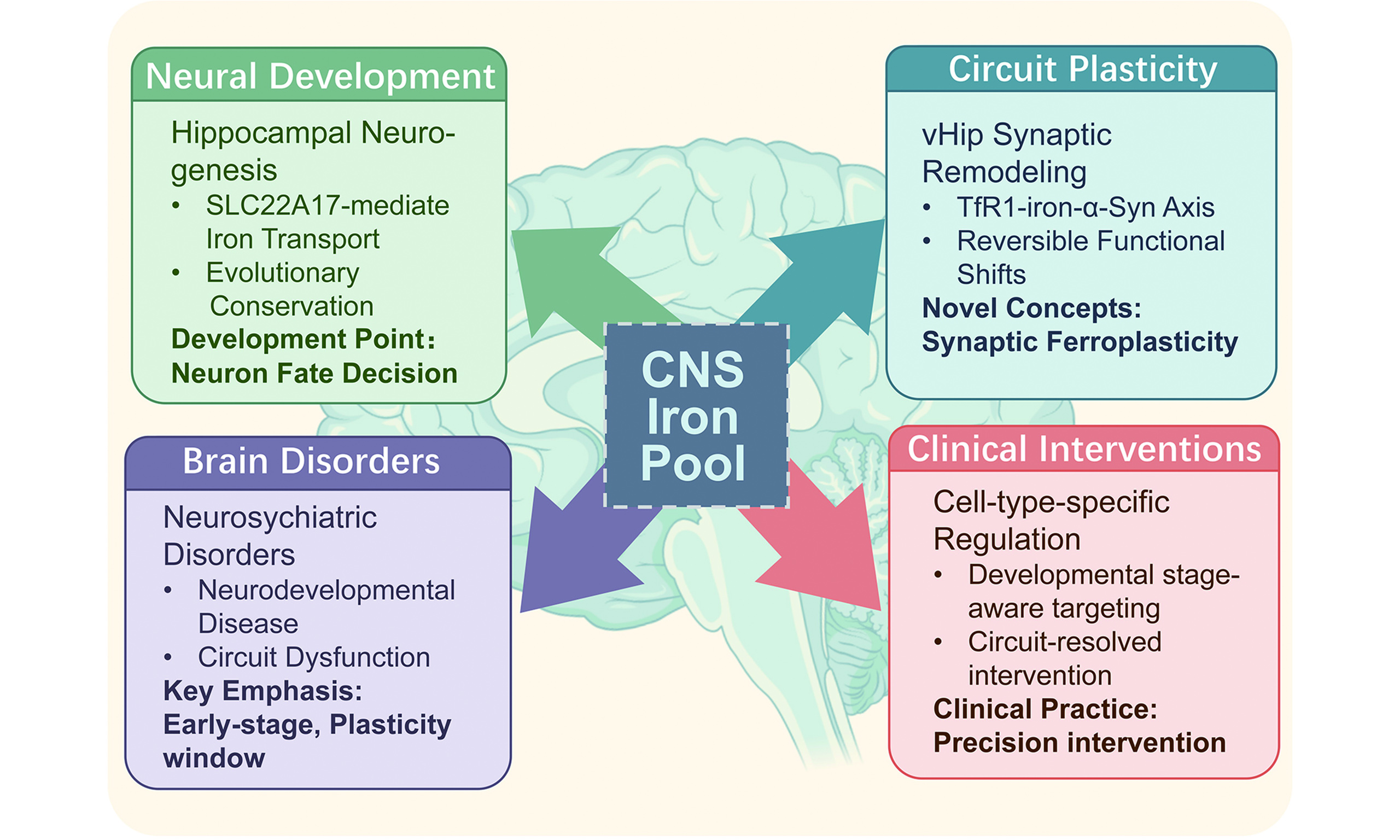
Highlights
1. Brain iron functions as an active regulatory trigger rather than merely an oxidative burden.
2. Iron homeostasis critically regulates neural fate determination and synaptic remodeling.
3. Specific regulatory axes, such as SLC22A17 and TfR1, directly modulate progenitor cell and synaptic dynamics.
4. The "ferroplasticity" framework conceptually links neurodevelopmental trajectories to psychiatric phenotypes.
5. Targeted brain iron modulation provides novel circuit-resolved and cell-type-specific therapeutic avenues.
INTRODUCTION: FROM FRAGMENTED INSIGHTS TO A UNIFYING FRAMEWORK FOR BRAIN IRON BIOLOGY
Iron is one of the most extensively studied metallic elements in the central nervous system (CNS). Research conducted in invertebrates, such as fruit flies, first established the role of iron in neurogenesis and development. However, studies in this field have failed to effectively integrate developmental and pathophysiological contexts within a coherent framework, instead focusing on isolated neurodegenerative diseases or discrete molecular pathways[1]. There remains a critical knowledge gap regarding how iron dynamics are spatiotemporally coordinated across the CNS throughout the lifespan. Recent advances in single-cell omics and neural circuit science have revealed the limitations of traditional models and necessitate a cross-scale reassessment. Our aim is to establish a unified framework that integrates molecular signaling with circuit-level function, thereby reconciling conflicting findings and establishing iron as a central, evolutionarily conserved regulator of CNS health and disease.
IRON HOMEOSTASIS ACROSS NEUROGENESIS: FROM METABOLIC SUPPORT TO FATE REGULATION
During the development of the CNS, the establishment of neuronal numbers and types is not accomplished in a single event, but unfolds progressively through a series of temporally and spatially tightly regulated events[2]. Upon entering the postnatal stage, restricted and region-specific neurogenesis persists in certain brain regions, including the subventricular zone (SVZ) of the lateral ventricle and the subgranular zone (SGZ) of the dentate gyrus in the hippocampus. During this period, neural stem cells and their progeny undergo a transition from self-renewal and fate selection to terminal differentiation, constrained by the local microenvironment. The focus of this developmental process starts to shift from cell number expansion to functional circuitry construction[2].
Neural stem cell differentiation involves significant state reorganization. Cell cycles shift, metabolism transitions from glycolysis to oxidative phosphorylation, and bioenergetic demands surge. Developmental regulation relies increasingly on metabolic states rather than just transcriptional or morphogenetic networks. Metabolic homeostasis elements thus become functionally critical. Iron, essential for electron transport, DNA synthesis, and enzymatic reactions, is increasingly recognized in neurogenesis[3].
Neurodevelopmental iron research has mainly focused on global homeostasis, overlooking the role of iron as a CNS developmental regulator. This interaction demands complex homeostasis for subtle regulation. A pioneering Nature Communications study[4] revealed solute carrier family 22 member 17 (SLC22A17; Slc22a17 in mouse)-mediated iron regulation continuously drives proper postnatal neurogenesis. Neural progenitor iron content dynamically rises and then declines from embryonic day 14 to postnatal day 14. This pattern correlates negatively with Slc22a17 expression. Nestin-Cre-driven Slc22a17 conditional knockout mice exhibit severe growth retardation, mortality, and impaired hippocampal neural stem cell proliferation and differentiation. These phenotypes suggest that SLC22A17 regulates cellular iron to maintain a hippocampal metabolic environment conducive to neurogenesis[4]. Disrupted regulation impairs differentiation rhythm and overall neurogenesis, and iron transcends mere nutritional support. It embeds within core cell fate decisions, co-shaping postnatal brain structural trajectories alongside traditional programs.
SLC22A17 research further elucidates mechanisms maintaining iron and redox homeostasis. As an endosomal membrane protein, SLC22A17 facilitates iron efflux via recycling pathways and binds sequestosome 1 (p62) to regulate the nuclear factor erythroid 2-related factor 2 (Nrf2), and heme oxygenase-1 (HO-1) pathway. In the absence of SLC22A17, abnormal p62 accumulation competitively binds Keap1, sustaining Nrf2 activation and HO-1 upregulation. Consequently, excessive HO-1-mediated ferric iron release amplifies oxidative stress. Despite iron accumulation and cell death, the lack of ferroptosis marker [e.g., glutathione peroxidase 4 (GPX4)] alterations suggests classical apoptotic characteristics[5]. This evidence highlights a distinct CNS regulatory layer, indicating neurogenetic iron metabolism operates relatively independently from global systemic control [Figure 1].
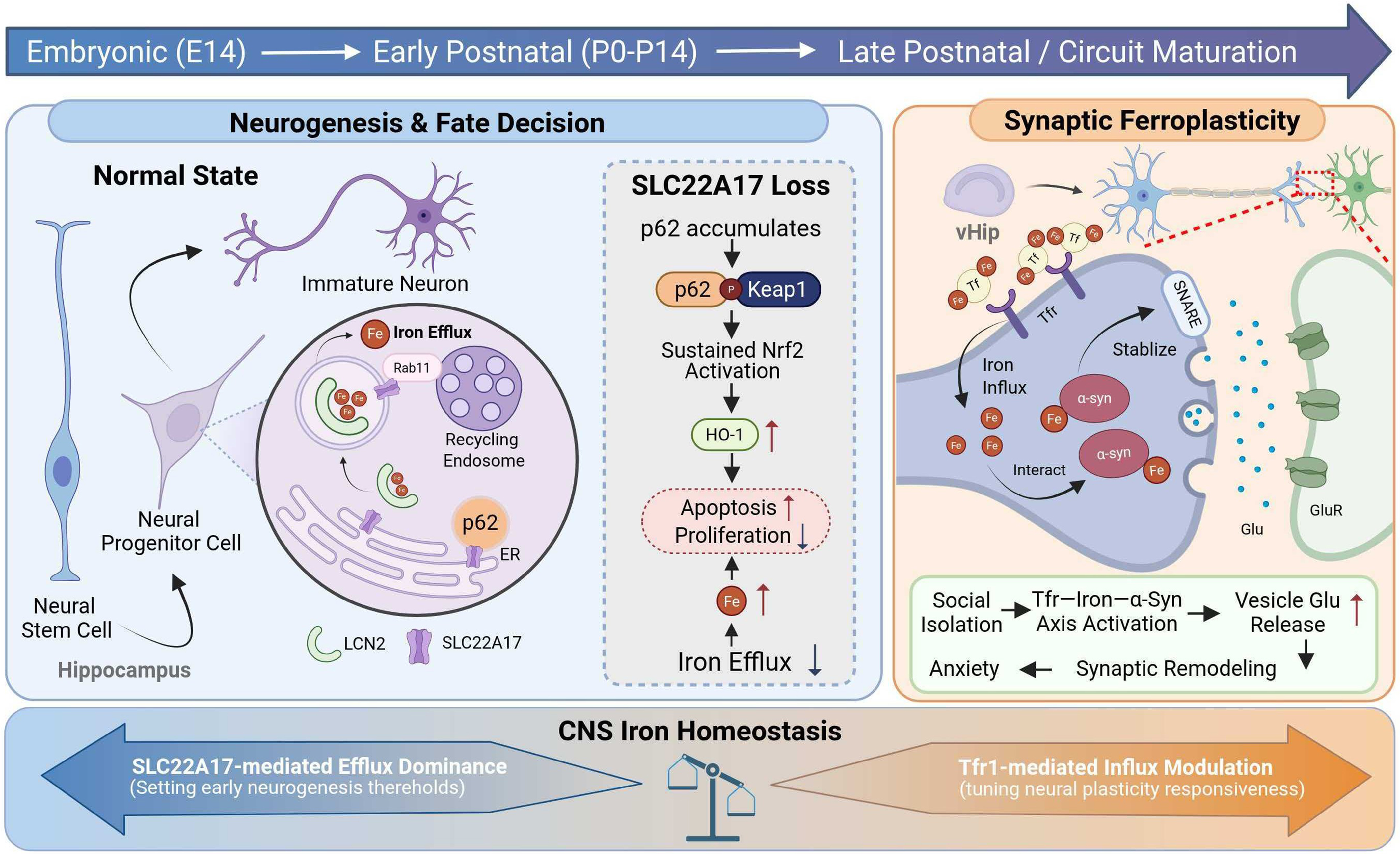
Figure 1. Iron as a dynamic regulator of neurogenesis and stress-induced plasticity. Iron plays an indispensable role throughout CNS neurodevelopment.
(1) During adult neurogenesis, neural stem cells and neural progenitor cells in the hippocampus regulate intracellular iron homeostasis via SLC22A17. SLC22A17-associated endosomal iron export contributes to the stabilization of cellular states and influences cell fate decisions during the neurogenic process; (2) Under conditions of social isolation, neurons in the vHIP exhibit increased TfR-mediated iron uptake. Through interactions with α-synuclein, iron modulates the stability of the SNARE complex, leading to enhanced presynaptic glutamate release. This process induces synaptic remodeling, reflected by an increased density of dendritic spines; (3) Across neurodevelopment, coordinated iron influx and efflux mediated by the SLC22A17/TfR1 axis contribute to the maintenance of brain iron homeostasis. CNS iron balance, in turn, exerts stage-dependent effects on multiple temporal windows of neurodevelopment [Created in BioRender. 2, 1. (2026) https://BioRender.com/6pg0954].
IRON HOMEOSTASIS DURING CIRCUIT MATURATION: FERROPLASTICITY AS A MECHANISM OF SYNAPTIC REMODELING
Once the initial fate of neurons has been determined, the focus of development shifts to their integration into functional networks; subsequently, structural remodeling and functional optimization at the synaptic level gradually become the primary drivers of neural circuit maturation. This stage involves the repeated formation, pruning and functional optimization of synapses. Neural circuits gradually transition from an initial state of high connectivity towards more specialized and efficient organizational forms[6]. This stage is highly dependent on energy metabolism, membrane synthesis and protein turnover, thereby establishing a close link between local metabolic states and synaptic structure and function.
A recent Cell Metabolism study[7] proposed this groundbreaking concept ferroplasticity in its research of the transferrin receptor 1 (TfR1)-iron-alpha-synuclein (a-Syn) axis in the ventral hippocampus. In contrast to previous research primarily linking iron imbalance to neurodegenerative disease, this research reveals that under specific environmental conditions, alterations in brain iron levels can directly modulate synaptic structure and function without inducing cell death. Within this framework, the authors identified a regulatory axis comprising glucocorticoid signaling, TfR1, and α-synuclein (α-syn), directly linking the environmental factor of social stress to alterations in iron homeostasis within the ventral hippocampus and anxiety-like behaviors[7]. Notably, similar to studies on SLC22A17, stress-induced iron accumulation does not accompany ferroptosis, further highlighting the unique nature of brain iron research.
At the mechanistic level, this study elaborates on the concept of ferroplasticity to describe a new form of synaptic plasticity contingent upon iron homeostasis. Specifically, the authors characterize this concept as a novel form of experience-dependent synaptic remodeling that targets ventral hippocampal pyramidal neurons through the activation of a glucocorticoid-initiated iron-α-Syn axis. This iron-dependent plasticity is not characterized as an aberrant pathway independent of classical synaptic plasticity mechanisms, but rather as a layer of modulatory logic embedded within and amplifying existing synaptic regulatory systems[7]. This work further advances research on brain iron from pathological associations towards mechanistic integration. Within the context of CNS neural development, this perspective establishes a foundation for re-examining the functional role of iron during synaptic remodeling. It also suggests that iron metabolism may constitute a crucial mediating layer linking environmental exposure, circuit plasticity, and affective disorders during the late stages of CNS neural developmental maturation [Figure 1].
SHIFTING DISEASE PARADIGMS: FROM NEURODEGENERATIVE DISORDERS TO NEURODEVELOPMENTAL AND PSYCHIATRIC CONDITIONS
Throughout the developmental journey of brain iron research, the theoretical framework and awareness of the issue remained largely restricted to neurodegenerative diseases for a long time. Research focus centered on how increased iron deposition, amplified oxidative stress, impaired mitochondrial function, and abnormal accumulation of harmful proteins such as α-synuclein and amyloid β, together with impaired clearance mechanisms, drive neuronal damage and ultimately lead to cellular death. This research pathway has accumulated substantial landmark findings, progressively establishing iron’s pathological role in conditions such as Parkinson’s disease (PD) and Alzheimer’s disease (AD)[8]. It has formed a relatively stable narrative logic. Iron acts as a potent pro-oxidant, amplifying oxidative damage and cellular dysfunction in neurodegenerative processes. Building upon this understanding, intervention strategies centered on chelating iron, limiting iron load, or inhibiting iron-dependent oxidative damage have emerged, constituting the early translational focus of brain iron research in disease contexts[9].
However, confining iron’s role in brain disorders solely to that of a ‘toxic factor’ clearly fails to encompass its broader biological impact. As research perspectives shifted from end-stage pathology towards developmental and functional stages, mounting evidence began pointing to the potential role of iron homeostasis imbalance in neuro-developmental abnormalities and psychiatric disorders mediated by neural circuit dysfunction[10]. This shift represents more than just an expansion of the disease spectrum. It also involves a repositioning of iron’s biological status in the brain.
Research on SLC22A17 reveals that iron homeostasis decisively influences progenitor cell fate and neuron generation timing[4]. Epidemiological and genetic evidence links perinatal iron deficiency and metabolic mutations to altered cognitive, emotional, and behavioral phenotypes, including developmental delays and autism-like behaviors[11]. Rather than a mere background metabolic variable, iron functions as a threshold regulator shaping critical neurodevelopmental decisions. Furthermore, ferroplasticity studies show that iron directly modulates synaptic plasticity and network activity without inducing neuronal death, altering neural circuit output[7]. These alterations manifest as emotional dysregulation or behavioral shifts, offering an explanatory pathway for psychiatric disorders distinct from structural damage. Mendelian randomization further links subcortical iron levels to conditions like major depression and schizophrenia[10]. The role of iron in brain disorders shifts from a pathological marker to a developmental and functional regulator, redefining disease essence from irreversible neuronal loss to functional deviations and regulatory imbalances within plasticity windows.
FROM OXIDATIVE BURDEN TO REGULATORY TRIGGER: REPOSITIONING IRON IN BRAIN BIOLOGY
How should iron’s role in brain biology be positioned? Traditionally, brain disease research frames iron around oxidative stress. As a Fenton reaction substrate, it explains reactive oxygen species (ROS) amplification, lipid peroxidation, mitochondrial dysfunction, and subsequent neuronal death[12]. This established a neurodegenerative pathological chain, cementing the dominant perception: the biological significance of iron lies primarily in its toxic excess. Iron typically enters the research spotlight only during pathological damage.
However, this masks the foundational metabolic role of iron in normal brain function. Iron stabilizes mitochondrial electron transport chains and serves as a necessary cofactor for the tricarboxylic acid (TCA) cycle, fatty acid, and purine synthesis. In the nervous system, these processes transcend mere metabolic support, coupling tightly with neurogenesis and neuronal maturation. During these energy-demanding, temporally precise phases, subtle iron supply-demand alterations reshape metabolic flux, redox states and cell fate[13]. Iron homeostasis extends beyond preventing toxic accumulation, constituting a critical metabolic microenvironment for successful CNS development. Two distinct iron homeostasis imbalance pathways emerge: toxic imbalance corresponds to a neurodegenerative terminal state, while regulatory imbalance operates at influencing progenitor cell fate, neuronal maturation, and synaptic priming during neurogenesis and circuit maturation. Operating as a sub-toxic signaling trigger, regulatory imbalance utilizes discrete pathways - from Jumonji C (JmjC)-domain demethylases to mammalian target of rapamycin complex 1(mTORC1) or p62-Nrf2 - to drive adaptive, reversible circuit shifts. Conversely, toxic imbalance breaches homeostatic buffering, reverting iron to raw chemical reactivity and triggering pervasive Fenton-mediated oxidative damage.
This bidirectional dysregulation imposes new research paradigm demands. Specifically, it requires clarifying the temporal window, cellular lineage, and circuit hierarchy where iron transitions from a regulatory variable to a pathological burden. Furthermore, determining whether this inflection point represents an intervention-eligible biological threshold is necessary. This provides a logical starting point for advancing from mechanistic understanding to precision intervention.
TOWARDS CIRCUIT-RESOLVED INTERVENTION: RETHINKING IRON REGULATION BEYOND SYSTEMIC HOMEOSTASIS
Translational therapies for iron and CNS disorders continue to face significant challenges. Clinical studies indicate that systemic iron chelation strategies yield limited efficacy in PD. Phase II clinical trials [SKY and EMBARK (ClinicalTrials.gov: NCT02728843; ANZCTR: ACTRN12617001578392)] featuring deferiprone demonstrated no consistent motor improvement in early-stage PD patients. Overall progression across different dosage groups was comparable to placebo, with some patients not receiving levodopa therapy even experiencing worsened symptoms. This was accompanied by a high incidence of adverse reactions[14]. These findings suggest that systemic iron reduction alone is insufficient to effectively intervene in CNS dysfunction.
Although the multifactorial nature of the disease contributes to treatment difficulty, an important factor is the highly spatiotemporal compartmentalization and cell-type specificity of central iron homeostasis, which are jointly determined by local metabolic demands, circuit activity states, and developmental stages. Therefore, precise regulation targeting specific brain regions, neuronal subtypes, and key iron transport pathways is emerging as a crucial direction in brain iron metabolism research. While the practical implementation of circuit-resolved intervention remains a significant challenge, several emerging technological platforms are already demonstrating transformative potential. For example, Cell-type-specific iron manipulation - achieved through Cre-loxP systems or viral-mediated gene editing - allows for the precise deletion or overexpression of key transporters such as SLC22A17 within defined neuronal or glial populations. Furthermore, advancements in functionalized nano-delivery systems offer a non-invasive avenue for delivering iron modulators to specific brain regions. These tools, combined with in vivo iron flux imaging, provide a clear path forward for transitioning from global iron management toward the restoration of regulatory precision within vulnerable neural circuits. In the meantime, cerebral iron homeostasis remains coupled with systemic metabolic states via liver-brain and gut-brain axes. Future intervention strategies must establish hierarchical coordination between systemic regulation and circuit-level targeting. This paradigm shift signifies a transition in cerebral iron therapeutics from holistic load management towards enhanced regulatory resolution[12].
CONCLUSION AND PERSPECTIVE
Viewing brain iron metabolism through neurogenesis and circuit plasticity reveals that its role in the CNS extends beyond basal demands. Iron homeostasis forms an evolutionarily conserved, autonomous regulatory module embedded in neural stem cell fate specification, synaptic remodeling, and circuit tuning. Emerging evidence shows that brain iron regulation operates in a cell-type- and context-dependent manner, dictated by neuronal subtype, developmental stage, and circuit activity, rather than passively extending systemic homeostasis. This paradigm shift elevates brain iron biology to an independent regulatory layer interfacing metabolic states, developmental programs, and plasticity. Three field-defining questions remain central to advancing this framework. First, what universal evolutionary principles govern the partitioning of iron flux between metabolic support and regulatory functions across CNS cell types and developmental epochs? Second, how do iron-dependent regulatory networks interface with neuroplasticity cascades to drive circuit adaptation, and what is the precise role of ferroplasticity in this crosstalk? Third, can we delineate molecular and circuit-level signatures that distinguish physiological iron modulation from pathogenic dysregulation to enable targeted interventions that preserve homeostatic function?
Within this framework, the limited efficacy of systemic iron interventions in CNS disorders - evidenced by iron chelator clinical trials[14] - is mechanistically clarified: it reflects a failure to account for the compartmentalized, cell-type-selective nature of brain iron dynamics. This underscores the imperative for circuit-resolved, cell-type-specific iron regulatory strategies tailored to distinct CNS regions and functional states. The bidirectional iron transport axis centered on SLC22A17 and TfR1 provides a paradigmatic molecular model for the dynamic calibration of iron flux across developmental and functional states. This framework offers a unifying basis for linking neurogenesis, ferroplasticity, and disease vulnerability, and lays a foundation for addressing the three core questions outlined above.
From a translational perspective, this framework extends beyond classical neurodegenerative paradigms to position iron dysregulation as a shared pathogenic factor across neurodevelopmental disorders, psychiatric conditions, and stress-related circuit dysfunction. These effects are mediated by subtle perturbations in developmental timing, synaptic plasticity, and network excitability, often occurring in the absence of overt neuronal loss. Future research must prioritize integrating iron metabolism into circuit-resolved disease models. Key emphasis should be placed on developmental windows of heightened vulnerability, selectively susceptible neuronal populations, and context-dependent regulatory thresholds that govern the transition from physiological iron homeostasis to pathological dysregulation. These directions directly address the three field-defining questions outlined above. Technological advances enabling cell-type-specific iron manipulation, in vivo iron flux imaging, and multi-omics integration will be pivotal to advancing this agenda[15].
Ultimately, repositioning iron as an active modulatory variable rather than a passive metabolic burden reframes therapeutic goals from global iron imbalance correction to restoring regulatory precision within defined neural circuits. This shift offers a nuanced, mechanism-driven path toward effective CNS disorder interventions and establishes brain iron biology as a cross-disciplinary cornerstone bridging neurodevelopment, metabolism, and psychiatry, thereby guiding the next generation of research.
DECLARATIONS
Authors’ contributions
Contributed to discussions, writing, and critical revision of all sections of the manuscript: Zhang, S.; Wang, Z.; Min, J.; Wang, F.; Chen, L.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (32130048 to Chen, L.; 32330047 to Wang, F.), the Ministry of Science and Technology of China National Key R&D Programs (2022YFA0806503, 2024YFA1306103 to Chen, L.), and the Starry Night Science Fund at the Shanghai Institute for Advanced Study, Zhejiang University (SN-ZJU-SIAS-0020 to Wang, F.).
Conflicts of interest
Wang, F. is the Editor-in-Chief of the journal Element. Min, J. is a Deputy Editor of the journal, and Chen, L. is an Editorial Board Member of the journal. Wang, F., Min, J. and Chen, L. were not involved in any steps of the editorial process, including reviewer selection, manuscript handling, or decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Paridaen, J. T.; Huttner, W. B. Neurogenesis during development of the vertebrate central nervous system. EMBO Rep. 15, 351-64 (2014).
3. Chen, L. et al. Homeostasis and metabolism of iron and other metal ions in neurodegenerative diseases. Signal Transduct. Target. Ther. 10, 31 (2025).
4. Tao, L. et al. Slc22a17 governs postnatal neurogenesis by maintaining the iron homeostasis in hippocampus. Nat. Commun. 16, 11117 (2025).
5. Zhao, J.; Wang, Y.; Tao, L.; Chen, L. Iron transporters and ferroptosis in malignant brain tumors. Front. Oncol. 12, 861834 (2022).
6. Faust, T. E.; Gunner, G.; Schafer, D. P. Mechanisms governing activity-dependent synaptic pruning in the developing mammalian CNS. Nat. Rev. Neurosci. 22, 657-73 (2021).
7. Wang, Z. et al. Ferroplasticity drives social isolation-induced anxiety via a ventral hippocampal iron-α-synuclein axis. Cell Metab. (2026). (Epub ahead of print).
8. Ayton, S.; Moreau, C.; Devos, D.; Bush, A. I. Iron on trial: recasting the role of iron in neurodegeneration. Brain 148, 4241-7 (2025).
9. Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms. Front. Pharmacol. 5, 99 (2014).
10. Du, W.; Tang, B.; Liu, S.; Zhang, W.; Lui, S. Causal associations between iron levels in subcortical brain regions and psychiatric disorders: a Mendelian randomization study. Transl. Psychiatry 15, 19 (2025).
11. McCarthy, E. K.; Murray, D. M.; Hourihane, J. O. B.; Kenny, L. C.; Irvine, A. D.; Kiely, M. E. Behavioral consequences at 5 y of neonatal iron deficiency in a low-risk maternal-infant cohort. Am. J. Clin. Nutr. 113, 1032-41 (2021).
12. Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron homeostasis and ferroptosis in human diseases: mechanisms and therapeutic prospects. Signal Transduct. Target. Ther. 9, 271 (2024).
13. Shapiro, J. S. et al. Iron drives anabolic metabolism through active histone demethylation and mTORC1. Nat. Cell Biol. 25, 1478-94 (2023).
14. Devos, D. et al.; a Parkinson’s disease study group. Therapeutic modalities of deferiprone in Parkinson’s disease: SKY and EMBARK studies. J. Parkinsons. Dis. 15, 72-86 (2025).
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
Article Notes
About This Article
Copyright







Data & Comments
Data
 0
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.