

Solvothermal synthesis and formation mechanism of lithium dodecaborate
 0
0 Abstract
Metal dodecaborates, particularly lithium dodecaborate (Li2B12H12), are promising ionic conductors, but their broader application is hindered by complex synthesis. Here, we report a facile solvothermal synthesis of Li2B12H12 via the reaction of lithium borohydride (LiBH4) with borane dimethyl sulfide complex (DMS·BH3) in glyme solvents. This synthesis can be conveniently performed either in a Schlenk flask (with or without reflux) or in an autoclave, demonstrating high yields (up to 96%) and excellent purity. The enclosed system provided by the autoclave was shown to be more favorable for the synthesis of the B12H122- anion. A detailed mechanistic investigation utilizing 11B nuclear magnetic resonance (11B NMR) spectroscopy revealed the stepwise formation of B2H7-, B3H8-, B9H14-, B11H14-, and B11H132- intermediates. This synthetic strategy was successfully extended to other alkali metal dodecaborates (Na, K), and their glyme-coordinated complexes were characterized by single-crystal X-ray diffraction. Furthermore, we introduce a solvent-exchange approach using weakly coordinating solvents such as dimethyl sulfoxide (DMSO) or water, enabling simple and efficient desolvation, thereby offering a practical new approach to obtain anhydrous metal dodecaborates.
Keywords
INTRODUCTION
The dodecahydro-closo-dodecaborate anion (B12H122-) has emerged as a versatile building block in advanced energy materials owing to its unique combination of properties[1-7]. This icosahedral cluster (theoretically predicted in the 1950s and first isolated in 1960) exhibits exceptional thermal and chemical stability and a highly delocalized charge, leading to the high ionic conductivity of its metal salts and enabling structural tunability through substitutions[8-14]. These properties have contributed to its increasing importance in a wide range of applications, notably as a superionic charge carrier in solid-state electrolytes, where recent studies have demonstrated fast lithium (Li)-ion transport, wide electrochemical stability windows, and improved safety compared to conventional liquid electrolytes[12,15-22], and as a boron-rich, water-soluble, and biocompatible agent with potential applications in high-energy materials, boron neutron capture therapy (BNCT), and various other functional systems[23-27]. However, the broader implementation of B12H122- is impeded by the lack of simple and scalable synthetic routes, which remains a major constraint on its broader application[3,4,11,28].
Conventional synthetic routes for B12H122- salts generally rely on either multi-step wet-chemical synthesis combined with ion exchange processes or solvent-free methods, as illustrated in Figure 1. In the classical solution-based route [Figure 1A], volatile borane precursors (e.g., B2H6, B10H14) react with borohydride anions in a high-boiling-point ethereal solvent[29]. To remove the strongly coordinating solvents, a widely adopted strategy involves an ion exchange step using amine complexes with bulky cations, typically yielding [(C2H5)3NH]2B12H12 as a water-insoluble intermediate. Subsequent ion exchange of this compound with metal hydroxides (or hydrides) allows for the preparation of the desired metal B12H12. Although this method yields hydrated metal dodecaborates and has been the preferred choice in earlier studies, its main drawbacks are operational complexity and high cost[30-32]. Solvent-free approaches [Figure 1B], based on ball milling or annealing, have gained popularity in recent years due to their ability to produce anhydrous B12H122- salts in a single step[33-35]. However, these approaches often result in the co-formation of other boron-rich compounds. In addition, both methods present concerns regarding product yield and purity, as well as the toxicity and reactivity of the specific borane precursors, which continue to pose significant challenges.
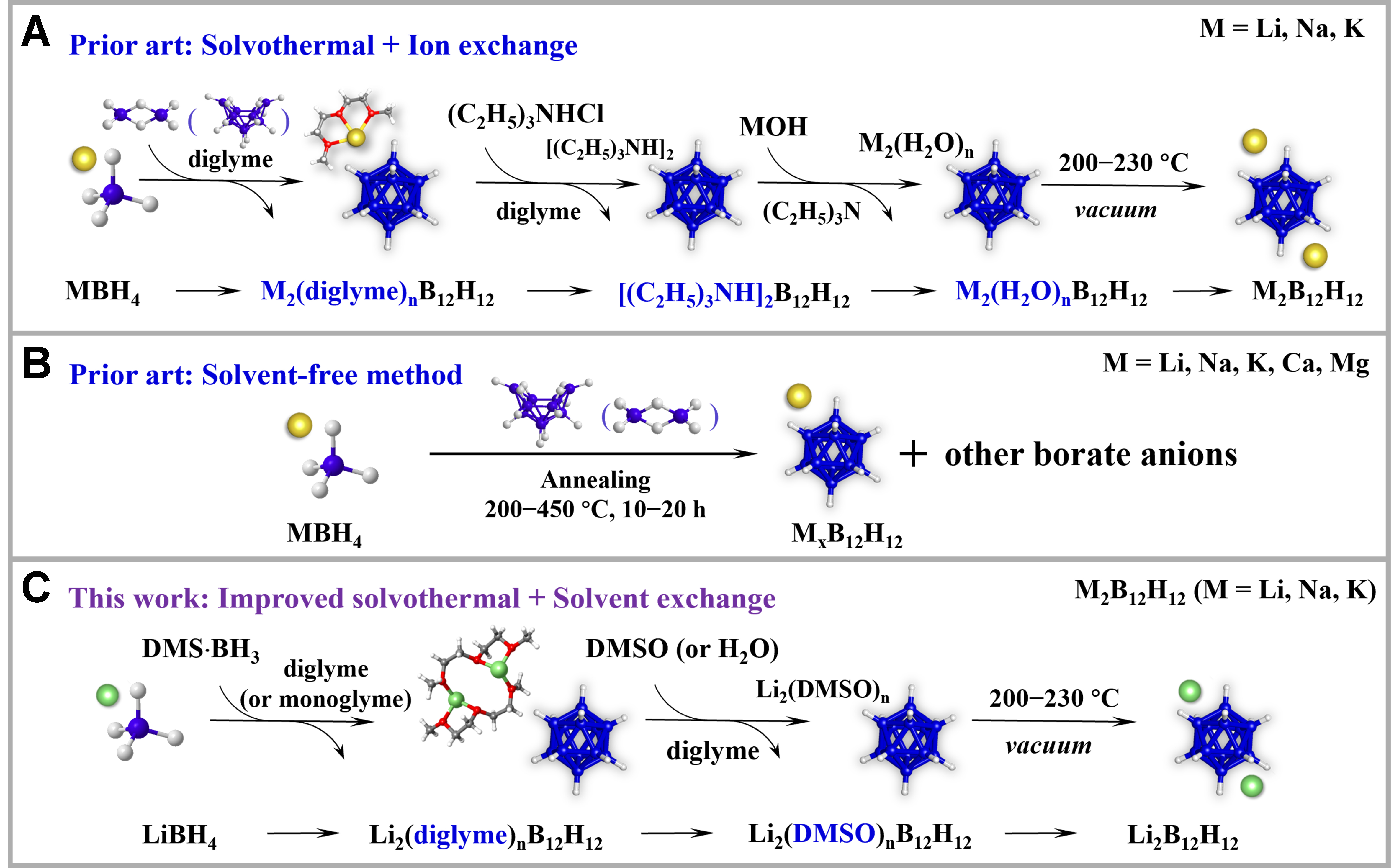
Figure 1. Comparative synthetic strategies for the preparation of metal dodecaborates. (A) Conventional solution-based synthesis followed by multi-step cation exchange; (B) Solvent-free synthesis; (C) This works: improved solvothermal synthesis followed by a solvent-exchange step. DMS·BH3: Borane dimethyl sulfide complex; DMSO: dimethyl sulfoxide; LiBH4: lithium borohydride; Li2B12H12: lithium dodecaborate.
Incremental improvements in synthetic approaches toward B12H122- have mainly focused on replacing diborane with reducing agents that present fewer handling issues[36-39]. With this in mind, we recently achieved the synthesis of unsolvated sodium and potassium dodecaborates (Na2B12H12 and K2B12H12) by reacting the borane dimethyl sulfide complex (DMS·BH3) with the corresponding borohydride (NaBH4 or KBH4) in diglyme under heating in an autoclave. The use of diglyme was found to facilitate the removal of all intermediate products formed during the reaction, leading to high-purity M2B12H12 (M = Na or K)[36]. Lithium dodecaborate (Li2B12H12) has recently attracted attention as an important precursor for producing Li-ion solid-state electrolytes[15-18,40]. In contrast to its sodium and potassium counterparts, limited research has been conducted on direct solution-phase synthetic approaches for obtaining this compound. Furthermore, due to the strong tendency of Li+ to coordinate with the ethereal solvents used in the synthesis, the production of unsolvated Li2B12H12 is particularly challenging[41,42]. One alternative approach involves exchanging Li+ with large monovalent cations, such as trimethylammonium, to yield water-insoluble products and enable the removal of diglyme [Figure 1A]. Anhydrous Li2B12H12 can then be obtained through a second cation substitution in the resulting [(C2H5)3NH]2B12H12 using LiOH in water, followed by heat treatment to readily remove the water[43]. However, this approach involves several steps and can introduce impurities if the amount of LiOH is not carefully controlled. In addition to the synthesis of B12H122-, cation exchange has been widely employed in the preparation of various metal borate compounds, including B11H14-, CB11H12-, and other metal borates[15,37,41,44]. These methods typically yield hydrated metal borates as intermediates, from which water can be removed under mild conditions to obtain the corresponding unsolvated salts. Such approaches often require multiple ion exchange steps and strict control of the exchange conditions. Therefore, a more straightforward and general strategy that avoids these complications would be highly desirable.
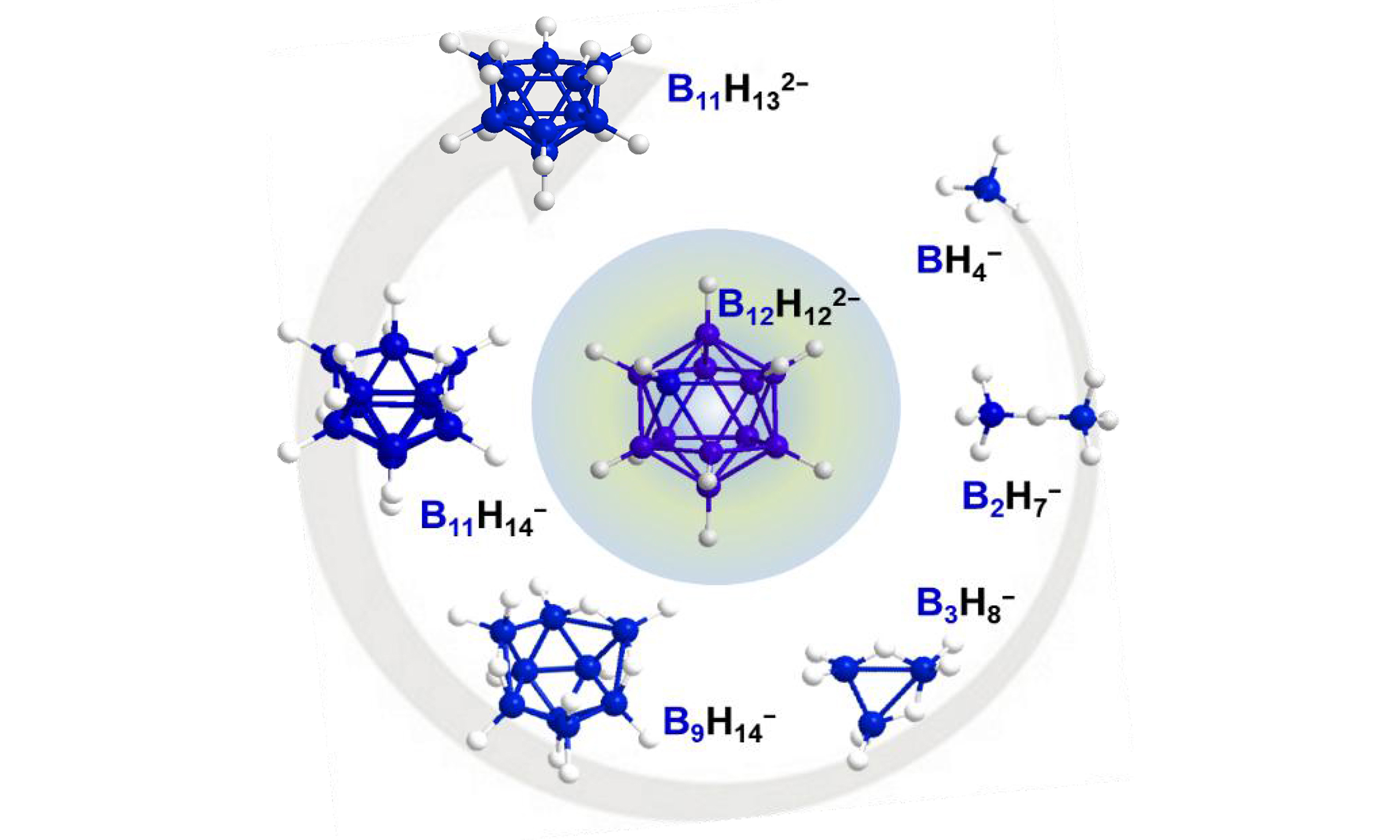
Since the observation of B12H122- formation as a “boron sink” during the dehydrogenation of BH4-, extensive experimental and computational work has been carried out to demystify the conversion process leading to B12H122-[34,37,45-49]. These studies demonstrated that BH4- can undergo a variety of pathways to form higher monovalent anions such as B3H8-, B9H14-, and B11H14-. The stepwise formation of larger polyhedral boranes from smaller boron clusters can be described as a series of reactions involving the incorporation of neutral boron hydrides, such as B2H6, into the BH4- anion. Indeed, small charged boron species, such as BH4- and B3H8-, tend to react with strongly electrophilic neutral boron hydrides, including L·BH3 (L = Lewis base), B2H6, B4H10, and B10H14, owing to their nucleophilicity[45,50,51]. However, the formation mechanism of the final dianion (B12H122-) from monovalent anions and neutral boron hydrides has not yet been well understood.
In this contribution, we present a novel and convenient synthetic approach [Figure 1C] for the preparation of highly pure anhydrous alkali metal dodecaborates, including Li2B12H12. The first step is based on reacting lithium borohydride (LiBH4) with DMS·BH3 in a heated solvent using either conventional Schlenk techniques [Supplementary Figures 1-3] or an autoclave. In the second step, a solvent exchange strategy coupled with thermal treatment is used to desolvate the product. Finally, the formation mechanism of B12H122- is experimentally investigated by solution-state 11B nuclear magnetic resonance (11B NMR) spectroscopy.
EXPERIMENTAL
Synthesis of Li2B12H12 in a Schlenk flask
Typically, 0.11 g of LiBH4 (5 mmol, 95%) was weighed into a glass frit-equipped Schlenk flask within a glovebox, and the flask was then sealed with a rubber septum before being removed from the glovebox. Subsequently, 20 mL of diglyme and 27.5 mmol (2.75 mL) of (CH3)2S·BH3 were added outside the glovebox using standard Schlenk techniques [Supplementary Figure 2A]. The solution was then heated to 120 °C, and the initially colorless solution turned yellow after approximately 2 h, accompanied by the formation of a white precipitate. The reaction mixture was further stirred at 120 °C for 24 h. The reaction solution was allowed to cool naturally to room temperature and was then filtered (using a 10-16 μm fritted glass filter) and washed with diglyme (3 × 5 mL). The resulting sample was dried under vacuum at 80 °C for 2 h to remove residual solvent. The solvated product, Li2B12H12·n C6H13O3, was obtained as a white crystalline solid with a yield of approximately 40% Li2B12H12 based on the amount of LiBH4 used.
Synthesis of Li2B12H12·n diglyme in an autoclave
Typically, 0.11 g of LiBH4 (5 mmol) was weighed into a quartz-lined reactor inside a glovebox and sealed with Parafilm®. Immediately before loading the quartz reactor into the autoclave, 20 mL of diglyme followed by 27.5 mmol (2.75 mL) of (CH3)2S·BH3 were quickly added to the borohydride. The autoclave was then sealed and purged with argon for 30 s to remove air from the system. The reaction temperature was increased from room temperature to 120 °C at a rate of 2 °C/min and maintained at this temperature for
Synthesis of Li2B12H12·n DME in an autoclave
Typically, 0.22 g of LiBH4 (10 mmol) was weighed into a quartz-lined reactor inside a glovebox and sealed with Parafilm®. Immediately before loading the quartz reactor into the autoclave, 20 mL of monoglyme followed by 55 mmol (5.5 mL) of (CH3)2S·BH3 were quickly added to the borohydride. The autoclave was then sealed and purged with argon for 30 s to remove air from the system. The reaction temperature was increased from room temperature to 120 °C at a rate of 2 °C/min and maintained at this temperature for
Solvent exchange of Li2B12H12·n diglyme with DMSO
Typically, 1 g of dried Li2B12H12·n diglyme was suspended in 10 mL of dimethyl sulfoxide (DMSO) in a glass frit-equipped Schlenk flask. Subsequently, the suspension was heated at 100 °C under vacuum to remove residual volatile solvents, affording Li2B12H12·n DMSO as a solid product. Final desolvation was achieved by thermal treatment at 200 °C for 12 h under dynamic vacuum, affording anhydrous Li2B12H12 as a white crystalline solid.
Solvent exchange of Li2B12H12·n DME with H2O
Typically, 1 g of dried Li2B12H12·n DME was suspended in 10 mL of deionized water in a glass frit-equipped Schlenk flask. Subsequently, the suspension was heated at 40 °C under vacuum to remove residual solvents, affording Li2B12H12·n H2O as a solid product. Final desolvation was achieved by thermal treatment at 200 °C for 12 h under dynamic vacuum, affording anhydrous Li2B12H12 as a white crystalline solid.
More experimental details are available in the Supplementary Materials.
RESULTS AND DISCUSSION
Synthesis of Li2B12H12 and its optimization
In the first step of our new synthetic method, LiBH4 is reacted with DMS∙BH3 in diglyme to produce Li2B12H12, following Equation (1). This reaction was first investigated using a classical Schlenk setup at 120 °C with a molar ratio of 1:5.5 (10% excess DMS∙BH3) [Supplementary Figure 4].
After 24 h, the yield of B12H122- in the solid product reached 45%, as shown by the sole presence of a doublet at around -15.2 ppm in the proton-coupled 11B solution-state NMR spectrum of the solid product dissolved in DMSO-d6 [Figure 2A][31,52,53]. NMR analysis of the filtrate [Figure 2B] revealed residual intermediates such as B3H8- and higher-nuclearity boron clusters, including B9H14-, B10H13-, and B11H14-, all of which are soluble in diglyme[42,54-56].
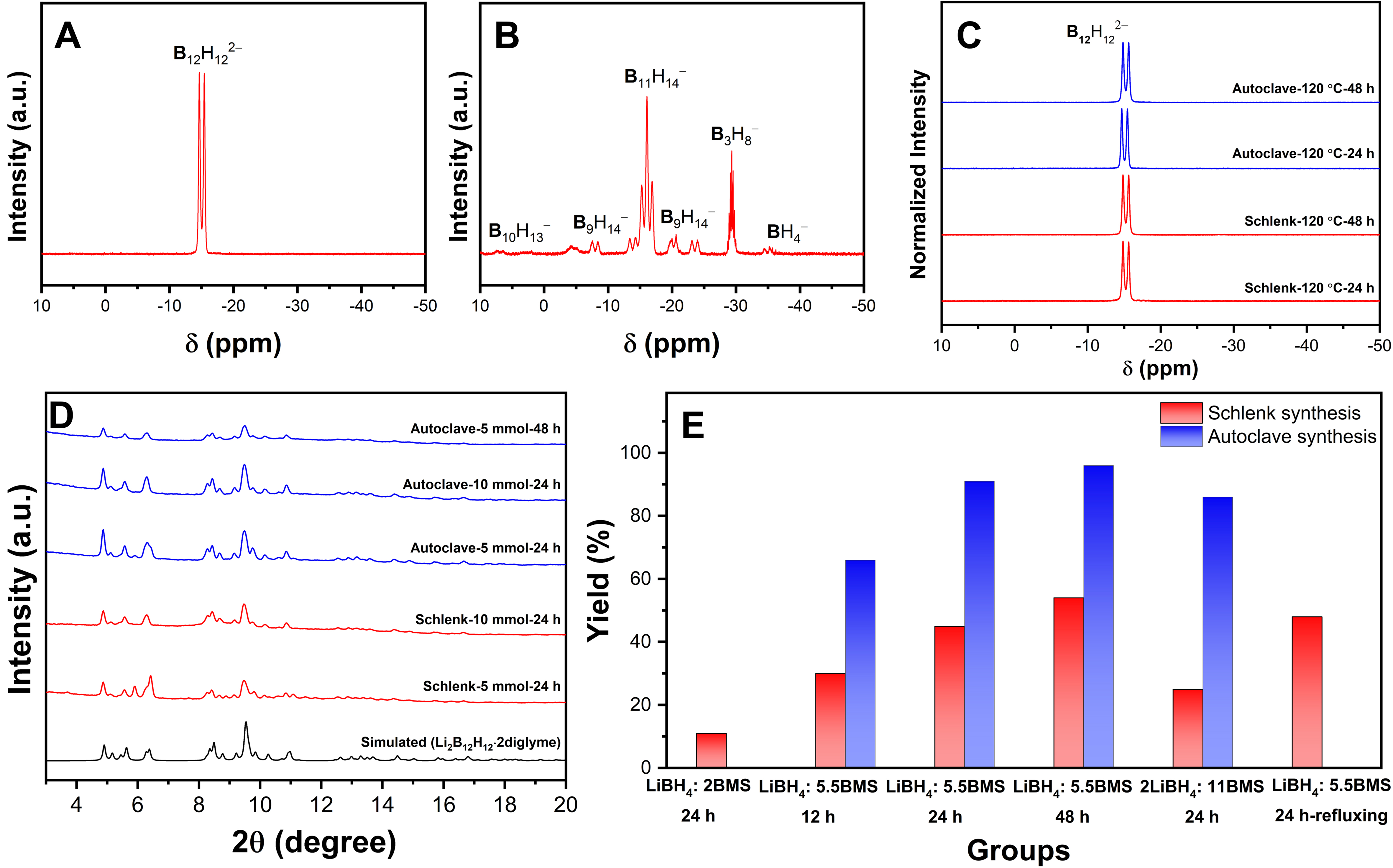
Figure 2. Proton-coupled 11B NMR spectra of reaction products: (A) solid precipitate and (B) filtrate after 24 h reaction at 120 °C in a Schlenk setup (5 mmol LiBH4 + 27.5 mmol DMS·BH3); (C) PXRD patterns of selected as-synthesized Li2B12H12·n diglyme samples (λ = 0.71073 Å); (D) Proton-coupled 11B NMR spectra of selected as-synthesized Li2B12H12·n diglyme samples; (E) Yields of selected as-synthesized Li2B12H12·n diglyme samples. 11B NMR: 11B nuclear magnetic resonance; LiBH4: lithium borohydride; DMS·BH3: borane dimethyl sulfide complex; PXRD: powder X-ray diffraction; Li2B12H12: lithium dodecaborate.
To optimize the reaction conditions, the DMS·BH3 ratio was varied while maintaining all other parameters constant (5 mmol LiBH4, 20 mL diglyme, and 24 h of heating at 120 °C). Experiments were carried out with LiBH4/DMS·BH3 molar ratios of 1:2 and 1:10. The 1:2 ratio, which is optimal for the synthesis of B3H8-, resulted in a Li2B12H12 yield of approximately 11% based on the amount of LiBH4 used, with B3H8- remaining as the main boron species in the filtrate [Supplementary Figures 4 and 5, Supplementary Table 2]. In contrast, the 1:10 ratio yielded 25% Li2B12H12 and produced B11H14- as the main boron species in the filtrate. Excess DMS·BH3 thus slightly increases Li2B12H12 yield but reduces BH3 utilization efficiency due to the formation of higher-nuclearity boron clusters (B9H14-, B11H14-), which deplete BH3 in the reaction mixture[55].
Temperature is a crucial factor in the synthesis of polyhedral borates, and its influence was therefore also investigated in this work[36,37]. At a lower temperature (85 °C), incomplete conversion of BH4- and DMS∙BH3 into B3H8-, B9H14-, B10H13-, and B11H14- was observed after 24 h, as shown by 11B NMR [Supplementary Figure 6], leading to a yellow reaction solution without precipitation. After extending the reaction time to 48 h, a very small amount of precipitate appeared, and the 11B NMR spectrum [Supplementary Figure 6] showed weak signals of B12H122- and near-complete depletion of LiBH4. At a higher temperature (160 °C) without a condenser, a poly-anionic species formed directly [Supplementary Figure 7]. We speculate that this species might arise from a reaction between B11H14- and partially dehydrogenated B12H12-x2- anions. Dehydrogenation of the icosahedral B12H122- cluster alters the charge and symmetry of the twelve BH vertices, leading to the formation of disubstituted anions when reacted with electrophilic compounds[57]. Performing the same experiment at 160 °C but using a setup equipped with a condenser resulted in the formation of B12H122-, along with some B10H102- and partially dehydrogenated B12H12-x2- anions [Supplementary Figure 8][58]. The use of a condenser also increased the yield compared to the reaction at 120 °C, as indicated in Supplementary Table 3. Additionally, the predominance of B11H14- in the filtrate [Supplementary Figure 9] indicates that refluxing promotes the conversion of lower boranes to B12H122-.
Similar reactions at 160 °C in Schlenk flasks equipped with a condenser [Supplementary Figure 10 and Supplementary Table 4], where LiBH4 was substituted by NaBH4 and KBH4, produced Na2B12H12 and K2B12H12 in isolated yields of 92% and 88%, respectively. In this case, higher temperature favors the formation of B12H122-, which is in line with previous research showing that the thermal decomposition of BH4- and B3H8- salts can generate closo-borates such as B10H102- and B12H122-[59-62]. Furthermore, in contrast to diglyme solvated Li2B12H12, the solvates of Na2B12H12 and K2B12H12 are much more stable at this temperature[36]. The small amounts of B10H102- impurities in Na2B12H12 and K2B12H12 can be completely removed by washing with diglyme. These results indicate that a Schlenk setup equipped with a condenser is suitable for high-yield synthesis of Na2B12H12 and K2B12H12 but is not ideal for Li2B12H12 preparation.
Further optimization of the synthesis conditions was performed by varying the concentrations of reactants and reaction times. The resulting products consistently exhibited high purity under all tested conditions, as shown in Figure 2C and D. The reactions were performed either in a Schlenk setup without a condenser or in an autoclave, as the latter enables all generated gaseous species, such as B2H6 and DMS, to remain confined within the reaction volume. As a general trend, the yields of Li2B12H12 obtained in the autoclave were much higher than those in the Schlenk setup. Based on the yield comparison presented in Figure 2E and Table 1, we speculate that B2H6 present in the autoclave atmosphere promotes the hydroboration reaction from BH4- to B11H14-, thus improving the yield of B12H122-[31,37,38,42,50,60,63]. Detailed reaction conditions are summarized in Supplementary Tables 3-7 and Supplementary Figures 11 and 12. In addition, longer reaction times enabled complete conversion of the reactants, resulting in higher yields. Remarkably, an isolated yield of 96% pure Li2B12H12 was achieved in the autoclave, representing, to the best of our knowledge, the highest yield achieved thus far for the preparation of metal dodecaborates [Supplementary Table 8]. When the reactant concentration in the autoclave was increased, a drop in yield was observed. This contrasts with the reactions in the Schlenk setup, for which increasing reactant concentrations or reaction times had no significant impact on the final yield of B12H122-.
Screening of synthetic conditions for Li2B12H12
| Entry (*) | Reaction setup | Reagents | Reaction conditions | Yield of Li2B12H12 (%) | ||
| LiBH4 (mmol) | DMS·BH3 (mmol) | Temperature (°C) | Time (h) | |||
| 1 | 5 | 10 | 120 | 24 | 11 | |
| 2 | Schlenk | 5 | 27.5 | 120 | 12 | 30 |
| Autoclave | 66 | |||||
| 3 | Schlenk | 5 | 27.5 | 120 | 24 | 45 |
| Autoclave | 91 | |||||
| 4 | Schlenk | 5 | 27.5 | 120 | 48 | 54 |
| Autoclave | 96 | |||||
| 5 | Schlenk | 10 | 55 | 120 | 24 | 40 |
| Autoclave | 86 | |||||
| 6 | 5 | 27.5 | 120 (Refluxing) | 24 | 48 | |
Signals of B10H13- were not observed in the 11B NMR spectra [Supplementary Figure 13] of filtrates resulting from autoclave experiments. The autoclave keeps all gaseous species under high pressure within the reactor system, and the B2H6 dimer can thus be retained in the reaction mixture. We speculate that the pyrolysis of B2H6 directly leads to the formation of higher boranes, such as B4H10 or B6H10, resulting in an increased content of B11H14- in solution and a slow reaction between B3H8- and B11H14-[64,65]. Remaining DMS·BH3 can be observed in the NMR spectra of products obtained from the autoclave system but not in those from reactions performed in a Schlenk flask.
The synthesis of Li2B12H12 in an autoclave can also be carried out using a lighter ether such as monoglyme (DME, boiling point of 85 °C) as the solvent. Supplementary Figures 14 and 15 show the 11B NMR spectra of the solid precipitate and filtrate obtained from the reaction of 10 mmol LiBH4 with 55 mmol DMS·BH3 in
Thermal behavior and desolvation of as-synthesized M2B12H12
After initial drying, the crude products contained variable amounts of diglyme molecules per formula unit, as evidenced by NMR spectroscopy [Supplementary Figures 11 and 12]. Attempts to remove coordinated solvent from Li2B12H12·n diglyme by thermal treatment were unsuccessful. Thermogravimetric analysis (TGA) was performed to investigate the thermal stability and composition of the Li2B12H12·n diglyme adducts [Supplementary Figure 16]. The thermal decomposition proceeds in three steps: the removal of free diglyme starts at 25 °C (-9.5 wt.%), followed by a larger mass loss of 15.5 wt.% starting at around 150 °C and a third mass loss of 5 wt.% between 200 and 300 °C. The latter two steps are attributed to the removal of coordinated diglyme.
An in situ synchrotron radiation powder X-ray diffraction (SR-PXRD) experiment was performed to further investigate the behavior of Li2B12H12·n diglyme during thermal treatment [Supplementary Figure 17]. The only crystalline phase observed in the diffraction patterns corresponds to Li2B12H12·2 diglyme, which is thermally stable up to 179 °C and then decomposes into an amorphous phase. The structure of Li2B12H12·2 diglyme was determined by single crystal X-ray diffraction (SC-XRD; Figure 3A, Supplementary Figure 59, Supplementary Table 9). In this solvate, two Li ions are threefold coordinated by three oxygen atoms from two diglyme molecules, evidencing strong interactions with the solvent molecules. It is noteworthy that upon reaching the decomposition temperature, the solid sample starts melting and bubbling. Hydrogen loss from the boron cages was confirmed by 1H NMR and 11B NMR [Supplementary Figures 18-20], as the characteristic asymmetric multiplet of B12H122- (1.6-0 ppm) disappears after thermal treatment at 170 °C under vacuum for 12 h, indicating competition between desolvation and dehydrogenation[66]. These results suggest that direct removal of diglyme by thermal treatment is not possible.
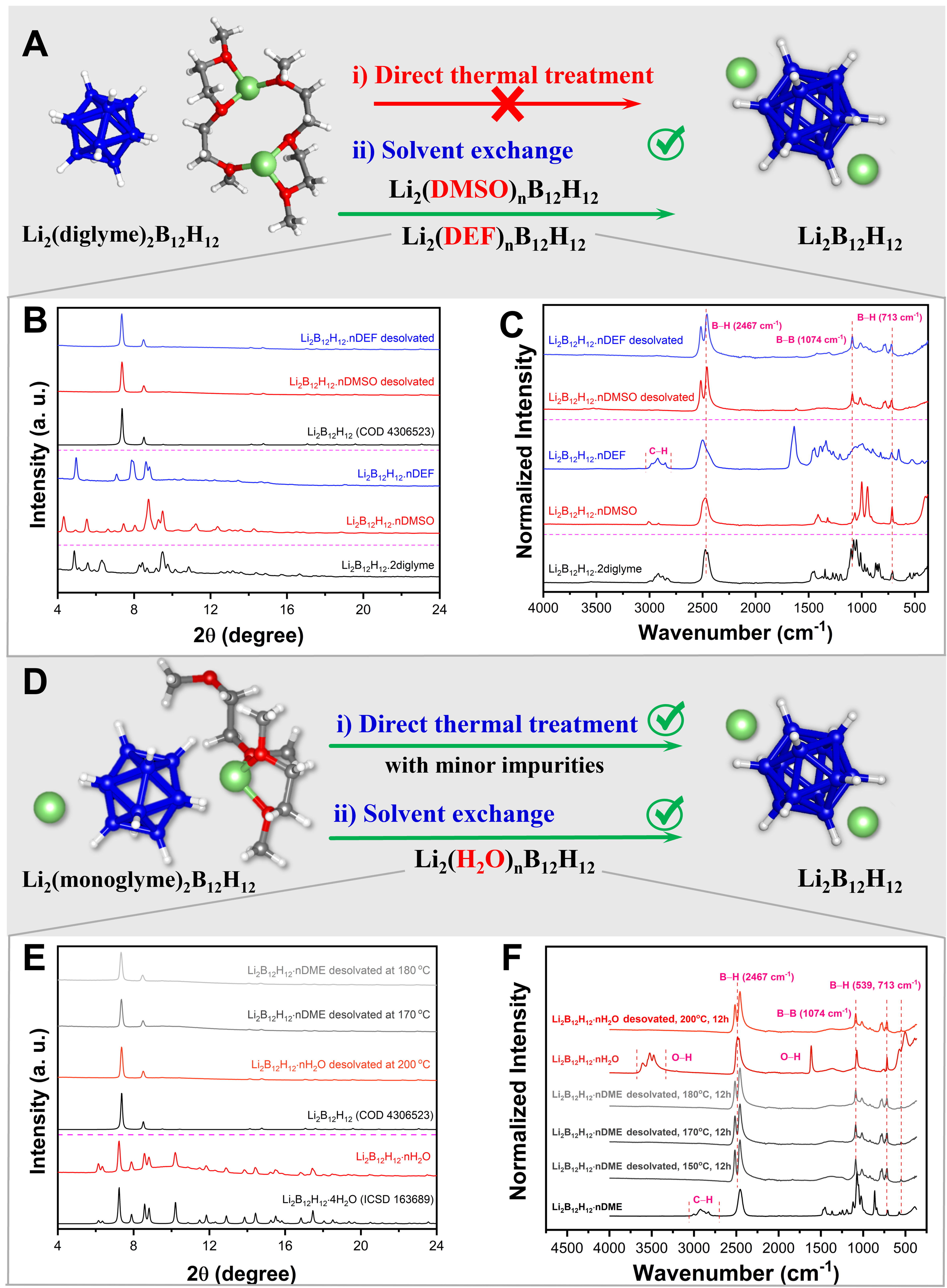
Figure 3. (A) Crystal structure fragment of Li2B12H12·2 diglyme and the approaches used for solvent removal; (B) PXRD (λ = 0.71073 Å) patterns; (C) FTIR spectroscopy spectra of Li2B12H12·n diglyme and DMSO/DEF-substituted samples before and after drying at 200 °C under vacuum; (D) Crystal structure fragment of Li2B12H12·1.5 monoglyme and the approaches used for solvent removal; (E) PXRD patterns (λ = 0.71073 Å); (F) FTIR spectra of Li2B12H12·n DME, Li2B12H12·n H2O, and anhydrous Li2B12H12 after heat treatment. Color code: Li (green), B (dark blue), H (light grey), O (red), C (dark grey). Li2B12H12: Lithium dodecaborate; PXRD: powder X-ray diffraction; FTIR: Fourier transform infrared; DMSO: dimethyl sulfoxide; DEF: N, N-diethylformamide; DME: 1,2-dimethoxyethane.
We investigated a new way to obtain anhydrous Li2B12H12 from glyme solvates, eliminating the need for the previously used and inconvenient ion exchange strategy, by using DMSO or N, N-diethylformamide (DEF) as solvent exchange agents. This approach aims to overcome the strong chelation of diglyme with dodecaborate salts containing small cations such as Li+ and Na+. The methodology consists of adding 10 mL of DMSO or DEF to 1 g of the diglyme-solvated product, followed by drying the solution under vacuum at 100 °C for 12 h. Upon this treatment, completely exchanged DMSO- or DEF-solvated Li2B12H12 is achieved, as demonstrated by powder X-ray diffraction (PXRD) and Fourier transform infrared (FTIR) spectroscopy [Figure 3A-C and Supplementary Figures 21-23]. The 1H NMR analysis [Supplementary Figure 24] of Li2B12H12·n DMSO before and after desolvation indicates that DMSO completely replaces diglyme. Additionally, DMSO can be removed from the solvate by thermal treatment without decomposition of the B12H122- anion. This suggests that DMSO can be used as an effective solvent for the removal of diglyme in Li2B12H12. Subsequently, drying the solvent-exchanged products at 200 °C under vacuum results in the formation of unsolvated Li2B12H12, as shown by in situ PXRD [Supplementary Figure 23] and NMR [Supplementary Figures 24-26]. The choice of DMSO and DEF for solvent exchange is justified by their higher boiling points and weaker coordinating ability compared to diglyme[67]. Although both solvents can effectively substitute diglyme, DMSO is the more economically viable option given the much higher cost of DEF.
When monoglyme is used in the synthesis, the crystalline phase of the obtained solvate corresponds to Li2B12H12·1.5 monoglyme, as evidenced by its structure determined by SC-XRD [Figure 3D, Supplementary Figure 60, Supplementary Table 10]. In this structure, monoglyme shows a coordinating ability toward Li+ similar to that of diglyme in solvated Li2B12H12, and thus may pose similar challenges for desolvation. However, in situ PXRD data [Supplementary Figure 27] reveal that monoglyme-solvated Li2B12H12 undergoes different phase changes during thermal desolvation compared to diglyme-solvated Li2B12H12, ultimately yielding Li2B12H12. Diffraction peaks of the monoglyme-solvated Li2B12H12 disappear at around 150 °C, while Li2B12H12 forms at this temperature. At 310 °C, a phase transition from the α-Li2B12H12 to β-Li2B12H12 is observed, followed by decomposition into a hydrogen-poor γ-Li2B12H12-x phase at higher temperatures[68,69]. This last phase transition occurs at a temperature approximately 50 °C lower than previously reported, likely due to the experiment being performed under vacuum. An attempt to directly remove monoglyme from the solvated Li2B12H12 by heating at 180 °C under vacuum for 12 h resulted in a small amount of Li2B12H12-x, as observed in the 11B NMR spectrum [Supplementary Figure 28].
Although complete removal of monoglyme from Li2B12H12 through direct heating is difficult, the solvent exchange strategy can in this case be implemented with an even lighter, more abundant, and greener solvent, namely water (H2O), compared to DMSO and DEF used for the diglyme-solvated dodecaborate. Indeed, upon exchange with water and applying vacuum at 40 °C, monoglyme can be completely removed, as shown in Figure 3D-F and 1H NMR spectra [Supplementary Figures 29-31], and pure hydrated Li2B12H12 can be obtained. Water can then be easily removed from hydrated Li2B12H12 without damaging the B12H122- clusters. It is worth noting that Li2B12H12 is commercially available in hydrated form, which can be conveniently obtained by our solvent exchange strategy without the last heating step.
Single crystals of diglyme-solvated Na2B12H12 and K2B12H12 were also isolated from the reaction mixtures during synthesis, and their structures were determined by SC-XRD to be Na2B12H12·4 diglyme [Figure 4A, Supplementary Figure 61, Supplementary Table 10] and K2B12H12·4 diglyme [Figure 4B, Supplementary Figure 62, Supplementary Table 11]. Unlike Li+, Na+ and K+ possess lower charge densities and larger cation sizes, resulting in higher cation-to-anion size ratios[70]. This leads to different coordination environments for Na+ and K+, with M+∙∙∙O bonds (Na+∙∙∙O = 2.378-2.446 Å, K+∙∙∙O = 2.728-2.844 Å) longer than Li+∙∙∙O = 1.958-2.039 Å, meaning that less energy is needed to break these coordination bonds. This finding explains why direct heat treatment of diglyme-solvated Na2B12H12 (150 °C) and K2B12H12 (200 °C) under vacuum effectively removed diglyme in our previous work, whereas the same approach was ineffective for the Li+ salt[36].
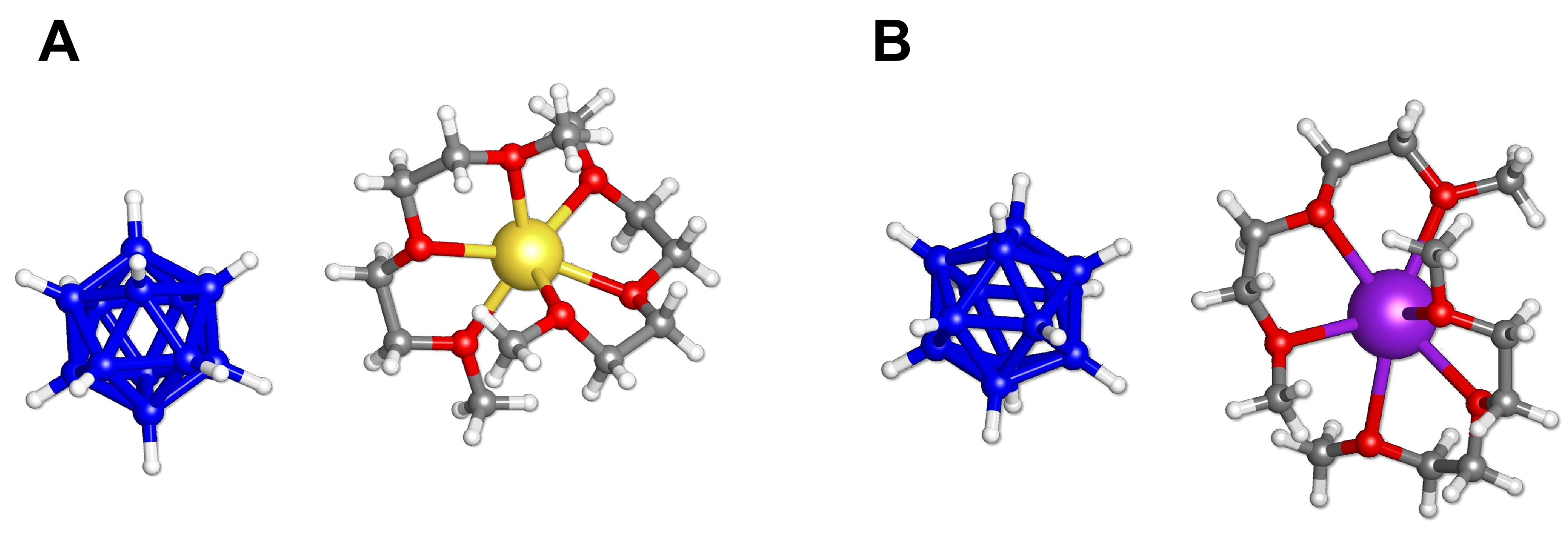
Figure 4. Fragments of the single-crystal structures of (A) Na2B12H12·4 diglyme (RT) and (B) K2B12H12·4 diglyme, illustrating the coordination modes of diglyme to alkali metal cations. Color code: Na, yellow; K, purple; B, dark blue; H, light grey; O, red; C, dark grey. RT: Room temperature.
Interestingly, drying crystals of diglyme-solvated Na2B12H12 at 80 °C for 2 h induced a single-crystal-to-single-crystal transformation, yielding a high-temperature (HT) diglyme-solvated form of Na2B12H12 [Supplementary Figure 63 and Supplementary Table 12]. Decomposition of the HT form, Na2B12H12·2 diglyme, directly yielded anhydrous Na2B12H12 [Supplementary Figures 32-36]. In contrast, decomposition of diglyme-solvated K2B12H12 proceeded steadily in a single step, leading to the formation of K2B12H12, as illustrated in Supplementary Figures 37 and 38.
We also attempted synthesis in toluene, a non-coordinating solvent, with the aim of directly obtaining unsolvated M2B12H12 (M = Li, Na). The 11B NMR spectra [Supplementary Figures 39-41] confirmed the formation of Li2B12H12 along with BH4-, B11H14-, B11H132-, and the thiomethyl-substituted cluster B12H11S(CH3)-[71]. PXRD analysis [Supplementary Figures 42-44] revealed Li2B12H12 as the only crystalline phase when the reaction was performed at 100 °C, while additional impurity peaks appeared at 120 °C. Performing the reaction in an autoclave at 120 °C achieved complete conversion of BH4- to B12H122-. We also ball-milled LiBH4 prior to synthesis in toluene to further improve the contact surface, as this reactant is insoluble in the solvent. This pretreatment significantly enhanced its reactivity, leading to the formation of BH4- and B11H132- together with B12H122- [Supplementary Figures 45-50]. While high-purity Li2B12H12 could be obtained from toluene, post-synthetic solvent extraction was necessary to remove residual byproducts. In contrast, replacing LiBH4 with NaBH4 primarily yielded NaB12H11S(CH3)2 rather than Na2B12H12
Elucidation of the formation mechanism of B12H122-
To gain insight into the mechanism of B12H122- formation, 11B NMR spectroscopy was employed to monitor the composition of the reaction mixture throughout the synthesis. Figure 5A shows the 11B NMR spectra of aliquots collected from a mixture of LiBH4 and DMS·BH3 in diglyme before heating (0 h) and at different time points during the solvothermal reaction at 120 °C.
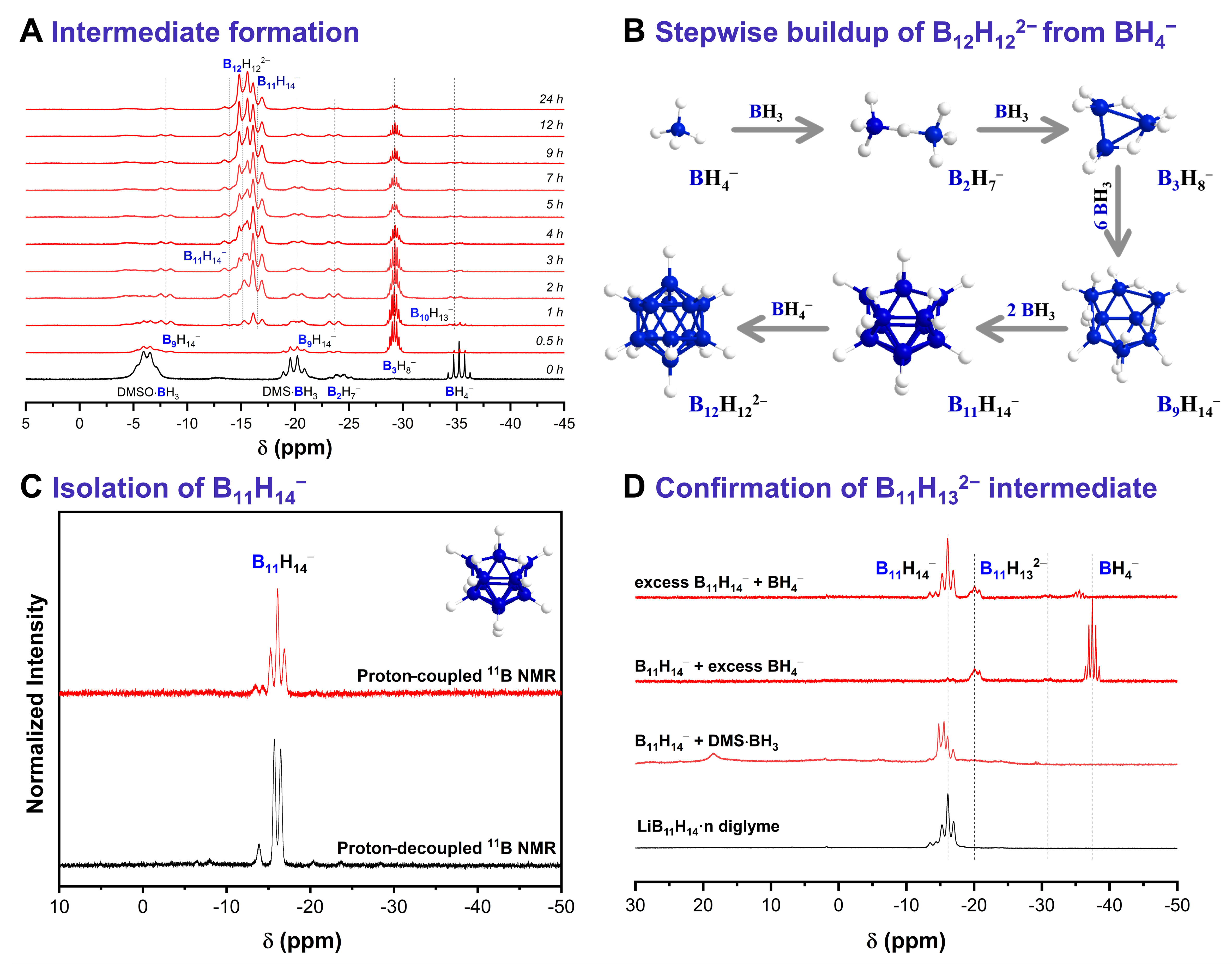
Figure 5. (A) Proton-coupled 11B NMR spectra of aliquots taken from a reaction mixture containing LiBH4 and DMS·BH3 in diglyme at
In the initial mixture at room temperature, a signal at -24 ppm was observed together with those of the reactants and was assigned to B2H7-[42]. This intermediate was then quickly converted into B3H8-, together with B9H14-, during the first half hour of the solvothermal reaction, in agreement with previous observations[42]. Notably, the intensities of the BH4- and DMS·BH3 signals decreased rapidly after initiating heating, accompanied by increasing concentration of B9H14- and B11H14- during the first hour of the reaction. This behavior is consistent with earlier reports on the dehydrocondensation of B3H8- with B2H6[72]. Furthermore, the characteristic doublet of B12H122- appeared after only 2 h of reaction, and its intensity increased continuously as the reaction progressed. In contrast, the intensities of the B9H14- and B11H14- signals remained almost constant throughout the reaction, while gradual consumption of B3H8- was observed. The sustained concentrations of B9H14- and B11H14- may indicate that these species act as intermediates in a dynamic equilibrium. Alternatively, their behavior could reflect a kinetic steady state in which the rates of formation and consumption are closely balanced, thereby preventing accumulation despite continuous turnover. Based on these 11B NMR observations, Figure 5B proposes a stepwise formation pathway for B12H122- involving the key intermediates B2H7-, B3H8-, B9H14-, and B11H14-.
The above experiment demonstrates that B3H8-, B9H14-, and B11H14- are stable intermediates in the sequential buildup of B12H122- from BH4- and BH3. To understand the final step in the formation mechanism of Li2B12H12, isolation of the intermediate B11H14- was critical. After several attempts [Supplementary Figures 53-55], this intermediate was successfully isolated by subjecting the reaction filtrate obtained from the initial synthetic step to thermal treatment at 160 °C [Figure 5C, Supplementary Figures 56 and 57]. To identify which species react with B11H14- to form dodecaborate, a solution of isolated LiB11H14 in diglyme was reacted with various lower boranes, including DMS·BH3 and BH4-, at 120 °C in a Schlenk flask for 3 h. As shown in Figure 5D and Supplementary Figure 58, direct reaction of LiB11H14 with BH4- did not yield B12H122-; instead, it produced B11H132- through deprotonation of B11H14- by BH4-. On the other hand, reaction of B11H14- with DMS·BH3 successfully generated B12H122-, whereas thermal decomposition of DMS·BH3 under the same conditions did not produce detectable amounts of B12H122- [Supplementary Figure 58]. Previous reports have demonstrated the isolation of the B12H122- anion through similar reactions, such as that between B11H14- and triethylamine borane at 150 °C. It has also been reported that Na2B12H12 can be obtained by reacting NaB11H14 with NaBH4 in boiling diglyme (boiling point = 162 °C)[8,37,39,41,55,73]. Our results indicate that at the lower reaction temperature of 120 °C, deprotonation is likely favored over direct hydroboration. Therefore, we propose that B11H132- serves as the terminal intermediate in this sequential pathway, and that its conversion to B12H122- proceeds only in the presence of sufficient BH3.
CONCLUSIONS
This work aims to develop a simple and efficient synthetic route to unsolvated Li2B12H12 with high purity and yield. A solvothermal strategy for Li2B12H12 synthesis based on the reaction of LiBH4 with DMS·BH3 in glymes was established, enabling the efficient synthesis of Li2B12H12 under optimized conditions with a 96% yield. An innovative methodology involving diglyme exchange enables the preparation of chemically pure Li2B12H12. The reaction intermediates formed during B12H122- synthesis were elucidated ex situ using 11B NMR. The results evidenced a stepwise buildup of B12H122- from BH4- through B2H7-, B3H8-, B9H14-, and B11H14- intermediates. The developed synthetic strategy for lithium and other alkali metal closo-dodecaborates, applicable to both solvated and unsolvated forms, represents a substantial advancement over previous approaches in terms of yield, purity, and cost-effectiveness. Beyond offering a practical route to M2B12H12 compounds, this work suggests a possible approach for the synthesis of other closo-borate materials that have traditionally relied on cation exchange.
DECLARATIONS
Acknowledgments
Dr. Dmitry Chernyshov and Dr. Iurii Dovgaliuk are acknowledged for their help during synchrotron measurements at the SNBL (Swiss-Norwegian Beamline) of the ESRF (European Synchrotron Radiation Facility) in Grenoble. The authors also acknowledge Dr. Igor E. Golub and Prof. Hans Hagemann (University of Geneva) for their meaningful discussions.
Authors’ contributions
Conceived and designed the study, conducted the experiments, performed data analysis and curation, prepared the figures, and wrote the original draft: Wang, J.
Contributed to the experimental work, data analysis, data validation, and manuscript revision: Steenhaut, T.
Carried out single crystal X-ray diffraction analysis, structure refinement, and structural data interpretation, and assisted in manuscript review: Robeyns, K.
Supervised the project, provided resources and funding, managed the research activities, and contributed to manuscript editing: Li, H. W.
Conceived the project, supervised the overall research, contributed to data interpretation, secured funding, and revised the manuscript: Filinchuk, Y.
Availability of data and materials
Detailed experimental procedures, characterizations, and supporting results are available from the Supplementary Materials. CCDC 2489735-2489739 contain the supplementary crystallographic data and can be downloaded free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was financially supported by the China Scholarship Council (201806930031), the FNRS (PDR T.0169.13, EQP U.N038.13, J.0164.17, CdR J.0073.20, and J.0168.22), and the Communauté Française de Belgique under Grant ARC 18/23-093.
Conflicts of interest
Li, H. W. and Filinchuk, Y. are Guest Editors of the Special Issue “Advanced Materials for Hydrogen and Energy Storage”. Li, H. W. and Filinchuk, Y. were not involved in any stage of the editorial process, notably including reviewer selection, manuscript handling, or decision making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Chen, C.; Chen, Z.; Zhang, M.; et al. Closo-[B12H12]2- derivatives with polar groups as promising building blocks in metal-organic frameworks for gas separation. ChemSusChem 2023, 16, e202300434.
2. Yan, Y.; Kühnel, R.; Remhof, A.; et al. A lithium amide‐borohydride solid‐state electrolyte with lithium‐ion conductivities comparable to liquid electrolytes. Adv. Energy. Mater. 2017, 7, 1700294.
3. Chen, C.; Ding, Z.; Gao, Y.; et al. Recent advances in boron chemistry. Sci. China. Chem. 2025, 68, 3927-95.
4. Wang, L.; Jiang, Y.; Duttwyler, S.; Lin, F.; Zhang, Y. Chemistry of three-dimensional icosahedral boron clusters anions: closo-dodecaborate (2-) [B12H12]2- and carba-closo-dodecaborate(-) [CB11H12]-. Coord. Chem. Rev. 2024, 516, 215974.
5. Ould, D. M. C.; Menkin, S.; Smith, H. E.; et al. Sodium borates: expanding the electrolyte selection for sodium-ion batteries. Angew. Chem. Int. Ed. Engl. 2022, 61, e202202133.
6. Green, M.; Kaydanik, K.; Orozco, M.; et al. Closo-borate gel polymer electrolyte with remarkable electrochemical stability and a wide operating temperature window. Adv. Sci. 2022, 9, e2106032.
7. Neumolotov, N. K.; Selivanov, N. A.; Bykov, A. Y.; et al. New methods for preparation of the monofluorosubstituted derivative of the closo-borate anion [2-B10H9F]2-, its properties, and analysis of its reactivity. Russ. J. Inorg. Chem. 2022, 67, 1583-90.
8. Adams, R. M.; Siedle, A. R.; Grant, J. Convenient preparation of the dodecahydrododecaborate ion. Inorg. Chem. 1964, 3, 461.
9. Eberhardt, W. H.; Crawford, B.; Lipscomb, W. N. The valence structure of the boron hydrides. J. Chem. Phys. 1954, 22, 989-1001.
10. Pitochelli, A. R.; Hawthorne, F. M. The isolation of the icosahedral B12H12-2 ion. J. Am. Chem. Soc. 1960, 82, 3228-9.
11. Jin, M.; Xu, D.; Su, Z.; et al. Hydridoborate-based solid electrolytes for all-solid-state batteries. Adv. Mater. 2025, e07809.
12. Paskevicius, M.; Jepsen, L. H.; Schouwink, P.; et al. Metal borohydrides and derivatives - synthesis, structure and properties. Chem. Soc. Rev. 2017, 46, 1565-634.
13. Bukovsky, E. V.; Peryshkov, D. V.; Wu, H.; et al. Comparison of the coordination of B12F122-, B12Cl122-, and B12H122- to Na+ in the solid state: crystal structures and thermal behavior of Na2(B12F12), Na2(H2O)4(B12F12), Na2(B12Cl12), and Na2(H2O)6(B12Cl12). Inorg. Chem. 2017, 56, 4369-79.
14. Campos dos Santos, E.; Sato, R.; Kisu, K.; et al. Explore the ionic conductivity trends on B12H12 divalent Closo-type complex hydride electrolytes. Chem. Mater. 2023, 35, 5996-6004.
15. Zhou, C.; Yan, Y.; Jensen, T. R. Enhanced electrochemical performance of the Li2B12H12-Li2B10H10-LiBH4 electrolyte. ACS. Appl. Energy. Mater. 2023, 6, 7346-52.
16. Maltsev, A. P.; Chepkasov, I. V.; Oganov, A. R. Order-disorder phase transition and ionic conductivity in a Li2B12H12 solid electrolyte. ACS. Appl. Mater. Interfaces. 2023, 15, 42511-9.
17. Simonyan, H.; Zhong, L.; Green, M. M.; et al. Solvation environment and interface dynamics of Li2B12H12 and Li2B12F12 electrolytes uncovered by theory and operando optical and FTIR spectroelectrochemistry. ACS. Appl. Mater. Interfaces. 2024, 16, 70028-37.
18. Asakura, R.; Łodziana, Z.; Grissa, R.; Rentsch, D.; Battaglia, C.; Remhof, A. Unveiling solid-state electrochemical oxidation of LiBH4 and Li2B12H12 for high-voltage all-solid-state batteries. ACS. Appl. Energy. Mater. 2025, 8, 9637-45.
19. Zhou, C.; Grinderslev, J. B.; Skov, L. N.; et al. Polymorphism, ionic conductivity and electrochemical properties of lithium closo-deca- and dodeca-borates and their composites, Li2B10H10–Li2B12H12. J. Mater. Chem. A. 2022, 10, 16137-51.
20. Pang, Y.; Liu, Y.; Yang, J.; Zheng, S.; Wang, C. Hydrides for solid-state batteries: a review. Mater. Today. Nano. 2022, 18, 100194.
21. Grams, R. J.; Santos, W. L.; Scorei, I. R.; et al. The rise of boron-containing compounds: advancements in synthesis, medicinal chemistry, and emerging pharmacology. Chem. Rev. 2024, 124, 2441-511.
22. Bachman, J. C.; Muy, S.; Grimaud, A.; et al. Inorganic solid-state electrolytes for lithium batteries: mechanisms and properties governing ion conduction. Chem. Rev. 2016, 116, 140-62.
23. Barth, R. F.; Mi, P.; Yang, W. Boron delivery agents for neutron capture therapy of cancer. Cancer. Commun. 2018, 38, 35.
24. He, Z.; Xiong, J.; Zuo, Y.; et al. Efficient targeted regulation of the interfaces and bulk in inverted perovskite solar cells with a [closo-B12H12]2--based derivative. Adv. Mater. 2025, 37, e2414155.
25. Huang, Z.; Wang, S.; Dewhurst, R. D.; Ignat’ev, N. V.; Finze, M.; Braunschweig, H. Boron: its role in energy-related processes and applications. Angew. Chem. Int. Ed. Engl. 2020, 59, 8800-16.
26. Li, J.; Kim, J. S.; Fan, J.; Peng, X.; Matějíček, P. Boron cluster leveraged polymeric building blocks. Chem. Soc. Rev. 2025, 54, 4104-34.
27. Xu, X.; Deng, X.; Li, Y.; et al. Applications of boron cluster supramolecular frameworks as metal-free chemodynamic therapy agents for melanoma. Small 2024, 20, e2307029.
28. Sivaev, I. B.; Prikaznov, A. V.; Naoufal, D. Fifty years of the closo-decaborate anion chemistry. Collect. Czech. Chem. Commun. 2010, 75, 1149-99.
29. Miller, H. C.; Miller, N. E.; Muetterties, E. L. Synthesis of polyhedral boranes. J. Am. Chem. Soc. 1963, 85, 3885-6.
30. Hansen, B. R. S.; Paskevicius, M.; Li, H. W.; Akiba, E.; Jensen, T. R. Metal boranes: progress and applications. Coord. Chem. Rev. 2016, 323, 60-70.
31. Chen, W.; Wu, G.; He, T.; et al. An improved synthesis of unsolvated NaB3H8 and its application in preparing Na2B12H12. Int. J. Hydrogen. Energy. 2016, 41, 15471-6.
32. Moury, R.; Gigante, A.; Hagemann, H. An alternative approach to the synthesis of NaB3H8 and Na2B12H12 for solid electrolyte applications. Int. J. Hydrogen. Energy. 2017, 42, 22417-21.
33. He, L.; Li, H.; Hwang, S. J.; Akiba, E. Facile solvent-free synthesis of anhydrous alkali metal dodecaborate M2B12H12 (M = Li, Na, K). J. Phys. Chem. C. 2014, 118, 6084-9.
34. Remhof, A.; Yan, Y.; Rentsch, D.; Borgschulte, A.; Jensen, C. M.; Züttel, A. Solvent-free synthesis and stability of MgB12H12. J. Mater. Chem. A. 2014, 2, 7244-9.
35. Yan, Y.; Rentsch, D.; Battaglia, C.; Remhof, A. Synthesis, stability and Li-ion mobility of nanoconfined Li2B12H12. Dalton. Trans. 2017, 46, 12434-7.
36. Wang, J.; Steenhaut, T.; Li, H. W.; Filinchuk, Y. High yield autoclave synthesis of pure M2B12H12 (M = Na, K). Inorg. Chem. 2023, 62, 2153-60.
37. Jing, Y.; Wang, X.; Han, H.; et al. Selective synthesis of the B11H14- and B12H122- borane derivatives and the general mechanisms of the B-H bond condensation. Sci. China. Chem. 2024, 67, 876-81.
38. Han, H.; Liu, X. R.; Gao, Y. M.; et al. An improved method for the synthesis of M2[B12H12] (M = Na, K) and their formation mechanism. Inorg. Chem. 2024, 63, 13886-92.
39. Jing, Y.; Liu, X. R.; Wang, X.; et al. Facile synthesis of the dodecahydridododecaborate (B12H122-) from borane Lewis base adducts. Sci. China. Chem. 2025, 68, 1355-61.
40. Zhou, C.; Sun, H.; Wang, Q.; et al. Highly electrochemically stable Li2B12H12-Al2O3 nanocomposite electrolyte enabling A 3.8 V room-temperature all-solid-state Li-ion battery. J. Alloys. Compd. 2023, 938, 168689.
41. Souza, D. H. P.; Møller, K. T.; Moggach, S. A.; et al. Hydrated alkali-B11H14 salts as potential solid-state electrolytes. J. Mater. Chem. A. 2021, 9, 15027-37.
42. Chen, X. M.; Ma, N.; Zhang, Q. F.; et al. Elucidation of the formation mechanisms of the octahydrotriborate anion (B3H8-) through the nucleophilicity of the B-H bond. J. Am. Chem. Soc. 2018, 140, 6718-26.
43. Huang, Z.; Gallucci, J.; Chen, X.; et al. Li2B12H12·7NH3: a new ammine complex for ammonia storage or indirect hydrogen storage. J. Mater. Chem. 2010, 20, 2743-5.
44. Kisu, K.; Kim, S.; Shinohara, T.; Zhao, K.; Züttel, A.; Orimo, S. I. Monocarborane cluster as a stable fluorine-free calcium battery electrolyte. Sci. Rep. 2021, 11, 7563.
45. Johnson, S. I.; Demaria, J. M.; Ginovska, B.; Edvenson, G. M.; Hagemann, H.; Autrey, S. T. Exploring detailed reaction pathways for hydrogen storage with borohydrides using DFT calculations. Energy. Fuels. 2022, 36, 5513-27.
46. Pitt, M. P.; Paskevicius, M.; Brown, D. H.; Sheppard, D. A.; Buckley, C. E. Thermal stability of Li2B12H12 and its role in the decomposition of LiBH4. J. Am. Chem. Soc. 2013, 135, 6930-41.
47. Yan, Y.; Remhof, A.; Rentsch, D.; Züttel, A. The role of MgB12H12 in the hydrogen desorption process of Mg(BH4)2. Chem. Commun. 2015, 51, 700-2.
48. Sethio, D.; Daku, L. M. L.; Hagemann, H.; Kraka, E. Quantitative assessment of B-B-B, B-Hb-B, and B-Ht bonds: from BH3 to B12H122-. Chemphyschem 2019, 20, 1967-77.
49. Bhattacharyya, P.; Boustani, I.; Shukla, A. Why does a B12H12 icosahedron need two electrons to be stable: a first-principles electron-correlated investigation of B12Hn (n = 6, 12) clusters. J. Phys. Chem. A. 2021, 125, 10734-41.
50. Chen, X.; Liu, X. R.; Wang, X.; Chen, X. M.; Jing, Y.; Wei, D. A safe and efficient synthetic method for alkali metal octahydrotriborates, unravelling a general mechanism for constructing the delta B3 unit of polyhedral boranes. Dalton. Trans. 2021, 50, 13676-9.
51. Zhao, Q.; Dewhurst, R. D.; Braunschweig, H.; Chen, X. A new perspective on borane chemistry: the nucleophilicity of the B-H bonding pair electrons. Angew. Chem. Int. Ed. Engl. 2019, 58, 3268-78.
52. Avdeeva, V. V.; Malinina, E. A.; Sivaev, I. B.; Bregadze, V. I.; Kuznetsov, N. T. Silver and copper complexes with closo-polyhedral borane, carborane and metallacarborane anions: synthesis and X-ray structure. Crystals 2016, 6, 60.
53. Ďorďovič, V.; Tošner, Z.; Uchman, M.; et al. Stealth amphiphiles: self-assembly of polyhedral boron clusters. Langmuir 2016, 32, 6713-22.
54. Dunks, G. B.; Barker, K.; Hedaya, E.; Hefner, C.; Palmer-Ordonez, K.; Remec, P. Simplified synthesis of decaborane(14) from sodium tetrahydroborate via tetradecahydroundecaborate(1-) ion. Inorg. Chem. 1981, 20, 1692-7.
55. Yin, Y.; Yan, F.; Li, S.; et al. Nature-inspired strategy: novel borohydride-based solid electrolytes extracted from cathode-electrolyte interphase. Adv. Mater. 2024, 36, e2406632.
56. Siedle, A. R.; Bodner, G. M.; Todd, L. J. Studies in boron hydrides - V: assignment of the 11B NMR spectrum of the tridecahydro decaborate(1-) ion. J. Inorg. Nucl. Chem. 1971, 33, 3671-6.
57. Kultyshev, R. G.; Liu, J.; Meyers, E. A.; Shore, S. G. Synthesis and characterization of sulfide, sulfide-sulfonium, and bissulfide derivatives of [B12H12]2-. Additivity of Me2S and MeS- substituent effects in 11B NMR spectra of disubstituted icosahedral boron clusters. Inorg. Chem. 2000, 39, 3333-41.
58. Gigante, A.; Duchêne, L.; Moury, R.; Pupier, M.; Remhof, A.; Hagemann, H. Direct solution-based synthesis of Na4(B12H12)(B10H10) solid electrolyte. ChemSusChem 2019, 12, 4832-7.
59. Fu, H.; Wang, X.; Shao, Y.; et al. Synthesis of lithium octahydrotriborate and investigation on its thermal decomposition. Int. J. Hydrogen. Energy. 2016, 41, 384-91.
60. Chen, X. M.; Jing, Y.; Kang, J. X.; et al. Synthesis, formation mechanism, and structure of K[BH3S(CH3)BH3] and its application in preparation of KB3H8. Inorg. Chem. 2022, 61, 12828-34.
61. Mishchenko, A. M.; Nechayev, M. A.; Subota, A. I.; et al. A new convenient method for preparing tetrabutylammonium closo-dodecaborate. J. Org. Pharm. Chem. 2024, 22, 3-9.
62. Zheng, X.; Yang, Y.; Zhao, F.; Fang, F.; Guo, Y. Facile preparation and dehydrogenation of unsolvated KB3H8. Chem. Commun. 2017, 53, 11083-6.
63. Hwang, S. J.; Bowman, R. C.; Reiter, J. W.; et al. NMR confirmation for formation of [B12H12]2- complexes during hydrogen desorption from metal borohydrides. J. Phys. Chem. C. 2008, 112, 3164-9.
64. Chen, X. M.; Ma, N.; Liu, X. R.; et al. Facile synthesis of unsolvated alkali metal octahydrotriborate salts MB3H8 (M=K, Rb, and Cs), mechanisms of formation, and the crystal structure of KB3H8. Angew. Chem. Int. Ed. Engl. 2019, 58, 2720-4.
65. Kotlensky, W. V.; Schaeffer, R. Decomposition of diborane in a silent discharge. Isolation of B6H10 and B9H15. J. Am. Chem. Soc. 1958, 80, 4517-9.
66. Chen, X.; Liu, Y. H.; Alexander, A. M.; et al. Desolvation and dehydrogenation of solvated magnesium salts of dodecahydrododecaborate: relationship between structure and thermal decomposition. Chemistry 2014, 20, 7325-33.
67. Alvarez, S. Coordinating ability of anions, solvents, amino acids, and gases towards alkaline and alkaline-earth elements, transition metals, and lanthanides. Chemistry 2020, 26, 4350-77.
68. Paskevicius, M.; Pitt, M. P.; Brown, D. H.; Sheppard, D. A.; Chumphongphan, S.; Buckley, C. E. First-order phase transition in the Li2B12H12 system. Phys. Chem. Chem. Phys. 2013, 15, 15825-8.
69. Yan, Y.; Wang, H.; Zhu, M.; Cai, W.; Rentsch, D.; Remhof, A. Direct rehydrogenation of LiBH4 from H-deficient Li2B12H12-x. Crystals 2018, 8, 131.
70. Payandeh, S.; Rentsch, D.; Łodziana, Z.; et al. Nido‐hydroborate‐based electrolytes for all‐solid‐state lithium batteries. Adv. Funct. Mater. 2021, 31, 2010046.
71. Hamilton, E. J. M.; Jordan; Meyers, E. A.; Shore, S. G. One-step preparation of dimethyl sulfide substituted icosahedral boranes: the crystal and molecular structures of 1,7-(SMe2)2B12H10, 1,12-(SMe2)2B12H10, and [SMe3][B12H11(SMe2)]·MeCN. Inorg. Chem. 1996, 35, 5335-41.
72. Gavrilova, L. A.; Titov, L. V.; Petrovskii, P. V. Synthesis of Bu4NB11H14 by the reaction of tetrabutylammonium octahydrotriborate with diborane in diglyme. Russ. J. Coord. Chem. 2004, 30, 307-8.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.