
Novel insight into genetic impacts on neurodegenerative dementia
 0
0 Abstract
Dementia can be broadly categorized into neurodegenerative dementias and non-neurodegenerative forms. Neurodegenerative dementias include Alzheimer’s disease (AD), frontotemporal lobar degeneration (FTLD), neuronal intranuclear inclusion disease (NIID), dementia with Lewy bodies (DLB), Huntington’s disease (HD), and prion diseases. Genetic factors play a central role in the etiology of neurodegenerative dementias. In AD, heritability estimates range from 58%-79% for late-onset AD (LOAD) and over 90% for early-onset AD, with causal genes including APP, PSEN1, and PSEN2. LOAD is a complex polygenic disease. Genome-wide association studies have identified more than 70 susceptibility loci, among which APOE ε4 is the most established genetic risk factor; carriers of the APOE ε4/ε4 genotype are now considered genetically predisposed to AD. However, the known heritability of AD remains incomplete, with rare variants in dozens of genes contributing substantially to disease risk. FTLD often presents with behavioral and language impairments, with MAPT, C9orf72, and GRN being the most commonly implicated causal genes. DLB, which overlaps clinically with Parkinson’s disease dementia, shares genetic risk factors with both AD and PD, including APOE, BIN1, SNCA, and GBA. NIID is caused by abnormal NOTCH2NLC GGC repeat expansions, which correlate with disease phenotype and age of onset. HD results from abnormal CAG repeat expansions in the HTT gene. Prion diseases arise from variants in the PRNP gene, with M129V being a notable risk factor. These findings underscore the pivotal role of genetic factors in the pathogenesis of neurodegenerative dementias.
Keywords
INTRODUCTION
Dementia is a syndrome characterized by progressive cognitive impairments in multiple cognitive domains. With the aging of the global population, the number of dementia patients is projected to increase from 57.4 million in 2019 to 152.8 million by 2050, posing a substantial burden on society and families[1]. Clinically, dementia arises from various causes and is commonly classified into neurodegenerative and non-neurodegenerative types. Neurodegenerative dementia (NDD) includes Alzheimer’s disease (AD), dementia with Lewy bodies (DLB), frontotemporal lobar degeneration (FTLD), neuronal intranuclear inclusion disease (NIID), Huntington’s disease (HD), and prion diseases. Multiple risk factors contribute to the pathogenesis of NDD and can be broadly categorized as modifiable and non-modifiable[2]. Among the non-modifiable factors, genetic components are well established. Advances in sequencing techniques and bioinformatic analyses greatly expanded our understanding of the genetics underlying NDD[3]. In this review, we summarize recent genetic findings related to NDD.
AD
AD is the most common form of dementia in the elderly, accounting for approximately 60%-80% of all dementia cases. Based on the age of onset, AD is classified into early-onset AD (EOAD; onset < 65 years) and late-onset AD (LOAD; onset ≥ 65 years)[4]. Genetics play a significant role in AD, with heritability estimates of 58%-79% for LOAD and over 90% for EOAD[5]. The major causal genes include APP, PSEN1, and PSEN2. To date, 364 PSEN1 variants, 115 APP variants, and 90 PSEN2 variants have been identified
Compared to EOAD, LOAD is much more common and is considered a complex disease with a polygenic background. Dozens of risk genes contribute to its development, with APOE being the first identified. The APOE gene, located on chromosome 19q13.2, has three major alleles: ε2, ε3, and ε4[9]. Among them, APOE
Beyond APOE, genome-wide association studies (GWAS) have identified over 70 common low-risk susceptibility loci for AD [Supplementary Table 1]. Since 2009, more than 10 large-scale GWAS have been conducted. In 2009, two large GWAS identified PICALM, CLU, and CR1 AD risk loci[13,14]. In 2011, five additional AD loci were found using GWAS meta-analyses (ABCA7, MS4A6A/MS4A4E, EPHA1, CD33, and CD2AP)[15,16]. In 2013, the International Genomic Alzheimer’s Project (IGAP) confirmed nearly all previously reported loci - except CD33 - and identified 11 new susceptibility loci[17]. In 2019, a genome-wide association by proxy (GWAX) approach revealed several novel loci associated with AD risk[18]. That same year, Kunkle BW et al. reported five new genome-wide loci (IQCK, ACE, ADAM10, ADAMTS1, and WWOX), with ACE and WWOX specifically linked to increased AD risk[19]. In 2021, a large GWAX study replicated 31 LOAD-associated loci and identified seven novel loci in a cohort of 1,126,563 individuals[20]. In 2022, another large GWAS revealed 75 LOAD loci - including 42 novel ones - based on 111,326 AD cases and 677,663 controls[21]. The AD-associated loci are involved in amyloid plaque formation, neurofibrillary tangle development, cholesterol metabolism, endocytosis/phagocytosis, and immune responses[22]. However, many of these common loci have not been fully verified in the Chinese population[23]. Only a few GWAS conducted in China have identified novel susceptibility loci. In 2018, Zhou et al. revealed that GCH1 and KCNJ15 increased AD risk in eastern China[24]. In 2020, Jia et al. identified four novel loci (GLRX, CTC-278L1.1, CTD-2506J14.1, and CHODL) associated with AD risk in the Chinese population[25]. In 2024, Ge et al. discovered that KIAA2013, SLC52A3, TCN2, and EGFR contributed to AD risk in Southeast and Southwest China[26]. Chinese GWAS have also replicated associations within the APOE region, though replication of many other loci has been unsuccessful. Differences between Hispanic white and Chinese populations may elucidate the complex etiology of AD across ancestries. While Chinese-specific genetic architecture reveals potential therapeutic targets, functional annotation remains limited compared to European-centric databases. Cross-ancestry functional genomics is therefore urgently needed to unlock diagnostic and therapeutic paradigms.
Although GWAS have revealed dozens of loci implicated in AD, the heritability estimated from GWAS summary statistics is only 3.1%-7.1%[20]. The discrepancy between heritability estimates from twin studies and GWAS is referred to as “missing heritability”, which may be attributed to several factors, such as underexplored rare variants and the limited sample sizes of non-European populations[22]. To address this missing heritability, several strategies can be explored, such as analyzing rare and complex variants, establishing large-scale cohorts across diverse ancestries, and incorporating gene-environment interactions. With the development of next-generation sequencing (NGS) technologies, including whole-exome sequencing (WES) and whole-genome sequencing (WGS), an increasing number of rare risk variants for AD have been identified. Unlike common variants detected through GWAS, rare variants identified by NGS are low-frequent in populations but have strong effect sizes. In European populations, genes such as TREM2[27], PLD3[28], ABCA7[29], UNC5C[30], SORL1[31], PLCG2, ABI3[32], ATP8B4, and ABCA1[33] have been implicated in AD risk. In the Chinese population, rare variants in PDE11A[34], C7[35], ACAA1[36], ECE2[37], GSN[38], LMTK2, and CRB1[39] have been associated with AD risk [Figure 1]. These rare variants influence Aβ or tau metabolism and contribute to AD pathogenesis.
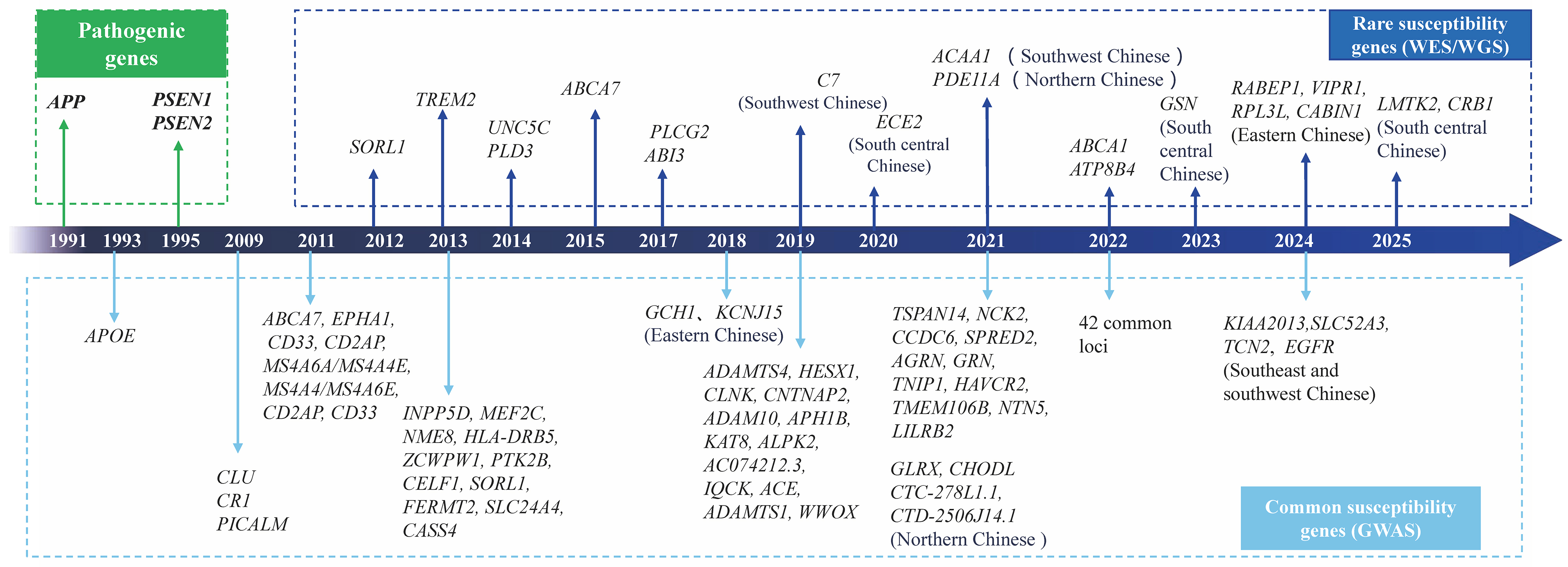
Figure 1. Pathogenic, common risk, and rare susceptibility genes associated with AD. Genes annotated with specific Chinese populations (e.g., Northern Chinese) represent novel associations first identified in our Chinese cohort. Genes without population annotations represent established risk genes originally discovered in European/American populations. AD: Alzheimer’s disease; WES: whole-exome sequencing; WGS: whole-genome sequencing.
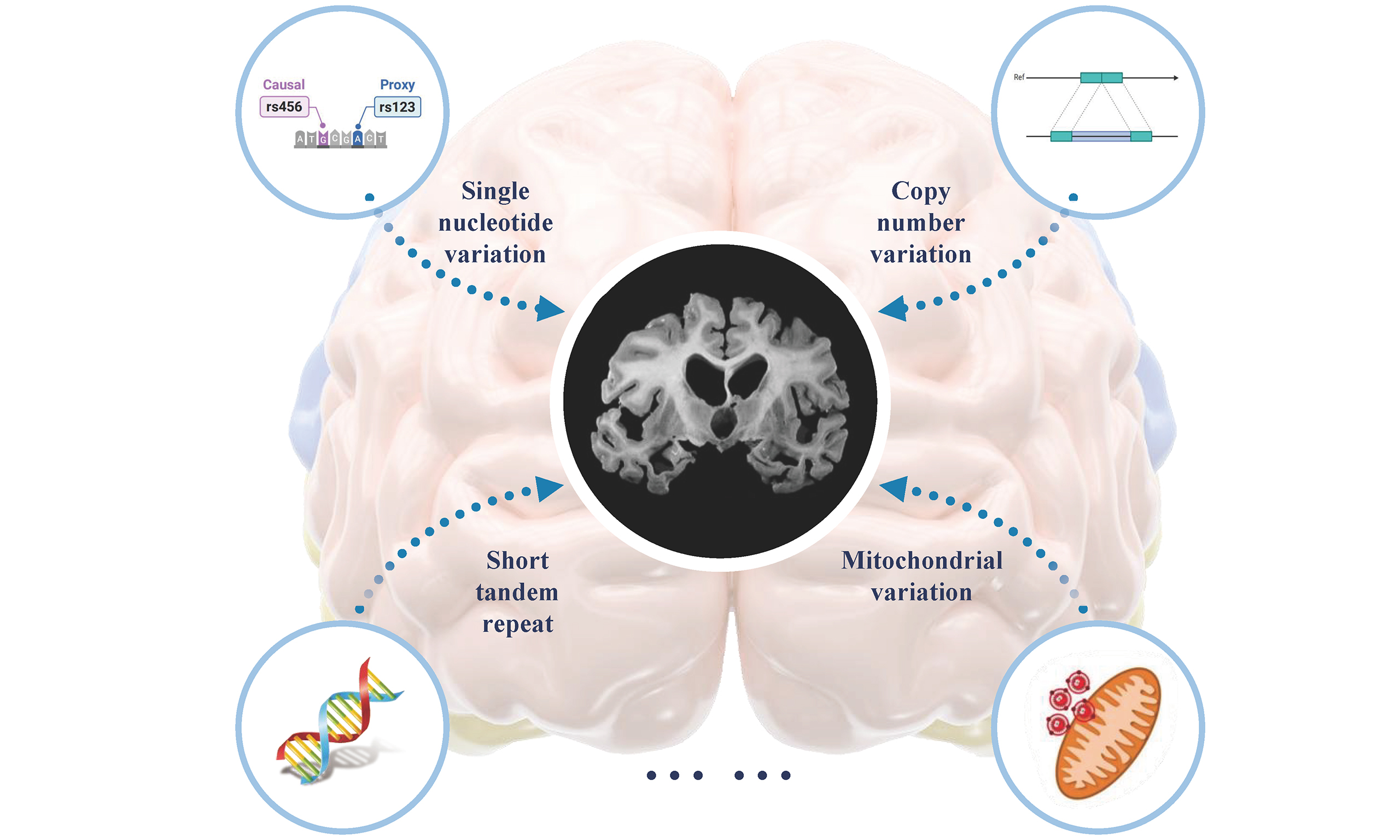
In addition to single nucleotide variants (SNVs) and insertions/deletions (indels), copy number variants (CNVs) have also been implicated in AD. CNVs are the most common type of structural variant, ranging in size from 50 bp to several Mb, and include duplications, deletions, insertions, inversions, and translocations[40]. It is estimated that 4.8%-9.5% of the human genome is affected by CNVs, which are associated with AD[41]. For example, 25 APP gene duplications have been shown to co-segregate with AD in autosomal dominant families[42]. In a study of 755 AD cases and 811 controls from non-Hispanic white, AD cases exhibited more duplications and larger deletions compared to controls[43]. Based on WGS data from 1,411 individuals, Chen Ming et al. identified 3,012 rare AD-specific CNVs enriched in biological processes such as cellular glucuronidation and neuronal projection. Integration of multi-omics data further revealed a key CNV potentially involved in immune response[44]. Tandem repeats represent another important type of genetic variation, consisting of DNA sequences repeated multiple times. Short tandem repeats (STRs) and variable number tandem repeats (VNTRs) are two major forms of repetitive sequences in eukaryotic genomes, with repeat units of 2-6 base pairs and 10-60 base pairs, respectively[45]. In Caucasian populations, Arne et al. found a strong correlation between VNTR length in ABCA7 and a GWAS signal. Expanded VNTRs were significantly enriched in AD patients, and VNTR length was negatively correlated with cerebrospinal fluid Aβ42 levels and ABCA7 expression[46]. In mainland China, NOTCH2NLC repeat expansions were identified in three clinically diagnosed AD patients[47]. Moreover, intermediate-length NOTCH2NLC CGG repeat expansions were also detected in a pathologically confirmed AD case[48]. These findings suggest that NOTCH2NLC CGG repeat expansions may be associated with AD. Beyond nuclear genes, mitochondrial variants also contribute to AD risk. In a cohort of 18,031 AD patients, the rare MT-ND4L variant rs28709356 was associated with AD onset. Furthermore, SKAT-O analysis implicated MT-ND4L and mitochondrial-related nuclear genes such as TAMM41 in AD pathogenesis[49].
FTLD
FTLD is one of the most common types of early-onset dementia, typically presenting between the ages of 45 and 65, and is characterized by progressive behavioral and/or language impairments. FTLD clinical syndromes include behavioral variant frontotemporal dementia (bvFTD), progressive non-fluent aphasia (PNFA), and semantic dementia (SD)[50]. PNFA and SD are classified as primary progressive aphasia (PPA). Among these, bvFTD shows the highest heritability, followed by FTD-ALS, PPA, and atypical Parkinson’s syndrome. The etiology and pathogenesis of FTLD are not fully understood, although genetic factors play a significant role. Around 20%-25% of FTLD cases carry a mutation associated with FTLD pathology, with autosomal dominant inheritance. To date, more than 10 pathogenic FTLD genes have been identified, with MAPT, C9orf72, and GRN being the most frequently implicated[51]. Over 50 pathogenic mutations in MAPT and more than 70 in GRN have been identified, accounting for 5%-20% and 5%-25% of hereditary FTLD cases, respectively[52,53]. C9orf72 variants account for 20%-30% of hereditary FTLD, with a pathogenic hexanucleotide repeat expansion (> 30 repeats) contributing to 25% of familial cases and 5% of sporadic cases in Caucasian populations[50,54].
MAPT, the first gene identified in FTD, is located on chromosome 17 and encodes the microtubule-binding tau protein[55]. Pathogenic MAPT variants can lead to predominantly 3R, 4R, or mixed 3R/4R tau. Missense variants usually disrupt tau’s ability to bind microtubules, while splicing variants alter the 4R-to-3R tau ratio[56]. C9orf72, located on chromosome 9, encodes a protein involved in endosomal transport and autophagy. In Asian populations, C9orf72 variants are extremely rare (0-4.8%)[57,58], but a recent large Chinese cohort study found that C9orf72 expansion is the most common one in FTLD, accounting for 8.2% of hereditary cases[59]. GRN, also located on chromosome 17, encodes progranulin (PGRN). Most pathogenic GRN variants are frameshift, splicing, or nonsense mutations that cause loss of function or haploinsufficiency[60]. Other less common FTLD genes include TBK1, VCP, CHMP2B, FUS, SQSTM1, TARDBP, CHCHD10, TIA1, CCNF, and CYLD [Figure 2], which contribute to FTLD through multiple pathways, such as autophagy and inflammation[50]. For example, CHCHD10 mutations occur in up to 7.7% of Chinese FTD patients, a frequency much higher than that in European populations (0.7%-2.6%)[61].
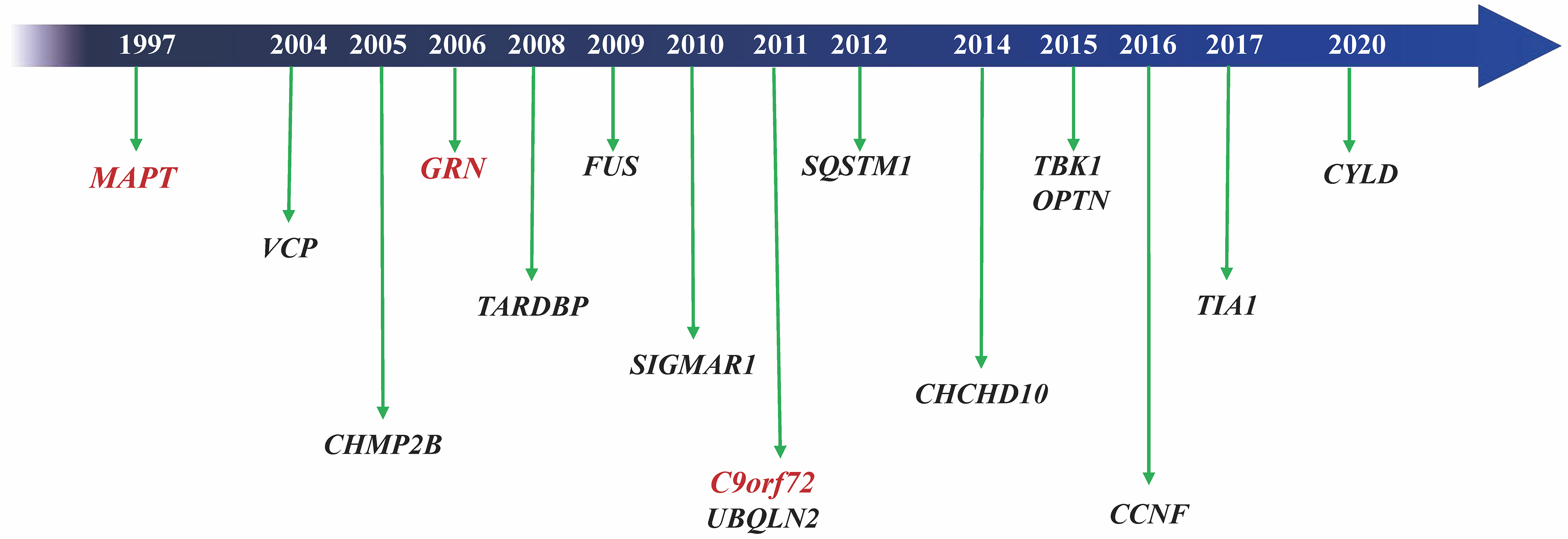
Figure 2. Pathogenic genes associated with FTD. FTD: Frontotemporal dementia.
In 2010, the first GWAS revealed that rs1990622 in TMEM106B was associated with reduced FTLD risk in 515 patients and 2,509 controls of Caucasian ancestry[62]. In 2014, a GWAS including 3,526 FTD patients and 9,402 controls showed a genome-wide significant association at the HLA locus and a suggestive association near RAB38/CTSC[63]. In 2019, a novel genome-wide significant risk locus (DPP6 rs118113626) was discovered using WGS data from 517 FTLD-TDP patients and 838 controls[64]. Rare variants also contribute to FTD risk. In a cohort of 2,139 FTD patients and 9,047 controls, a rare missense variant in MAPT (p.A152T) was associated with FTD risk[65]. Two rare variants near C9orf72 were strongly associated with FTD in 354 FTD patients and 4,209 controls[66]. Additionally, in European populations, GWAS have identified common CNVs in C9orf72 and MAPT associated with FTD-ALS. Rare structural variants, including LRRK2 duplication, CHCHD10 deletion, and FIG4 deletion, have been reported in patients with PPA, FTD-ALS, and FTD, respectively[67].
DLB AND PARKINSON’S DISEASE DEMENTIA
Lewy body dementias (LBD) include DLB and Parkinson’s disease dementia (PDD), which share substantial overlap in clinical features, genetics, and neuropathology[68]. DLB is the second most common NDD after AD, accounting for approximately 10%-20% of all dementia cases[69]. Genetic factors are estimated to contribute about 36% to the development of DLB. A GWAS including 1,743 DLB patients and 4,454 controls indicated that APOE, SNCA, and GBA loci were associated with DLB risk[70]. A larger GWAS confirmed these associations and additionally revealed BIN1 and TMEM175 as novel risk loci in 2,591 DLB patients and 4,027 controls[71]. The APOE ε4 allele is associated with faster disease progression and worsened survival in DLB[72]. APOE and BIN1 were linked to AD, while SNCA, GBA, and TMEM175 were associated with PD, highlighting the genetic overlap among DLB, AD, and PD. CNV also plays a role in DLB; in a European cohort, a common deletion in TPCN1 was identified as a novel risk locus[67]. Clinically, the “1-year rule” is often used to distinguish DLB from PDD: when dementia occurs more than one year after the onset of parkinsonian symptoms, the condition is more likely PDD; if cognitive symptoms precede or appear within one year of parkinsonism, DLB is more likely. Different variants in the SNCA gene are implicated in PDD, including missense variants and locus multiplications. In a cohort of patients with PD, PDD, and DLB, distinct associations were observed at the 3’ and 5’ regions of the SNCA gene, indicating that PD, PDD, and DLB may have partly distinct genetic etiologies[73]. Among 740 PD patients, carriers of GBA variants exhibited more rapid progression of cognitive decline during follow-up[74]. In another study of 37 PD patients and 40 controls, common variants in MAPT were associated with dementia in PD and with neural circuitry underlying cognition in controls[75]. Furthermore, a genome-wide survival analysis of 3,923 clinically diagnosed PD patients identified APOE ε4 and the APP receptor LRP1B as genetic risk factors for progression from PD to PDD[76].
NIID
NIID is a rare degenerative disease characterized by highly variable clinical symptoms and multisystem involvement. Based on predominant clinical features, NIID can be classified into four subtypes: dementia-dominant, movement disorder-dominant, muscle weakness-dominant, and paroxysmal symptom-dominant. Among these, the dementia-dominant subtype is the most common, present in 38.1% of patients and accounting for symptoms in 49.4% of cases[77]. Typically, patients with the dementia-dominant subtype exhibit subcortical dementia symptoms, such as early executive dysfunction and memory impairment[78]. In 2019, researchers in China and Japan identified abnormal GGC repeat expansions in the 5’ region of the human-specific NOTCH2NLC gene as the genetic cause of NIID. Pathogenic GGC expansions are defined by repeat sizes ranging from 66 to 517. A repeat size greater than 65 establishes a diagnosis of NOTCH2NLC-associated NIID[79,80]. Such pathogenic NOTCH2NLC GGC expansions are relatively common in Chinese NIID patients. However, they are rare among patients of European descent, indicating that additional genetic factors may contribute to NIID onset in these populations[81]. The size of the NOTCH2NLC GGC expansion is associated with specific clinical phenotypes. Larger expansions (200-517 repeats) are frequently observed in muscle weakness-dominant cases and are often characterized by a higher frequency of GGA interruptions and fewer AGC interruptions. Intermediate repeat sizes (100-200 repeats) are typically carried by patients with the dementia-dominant subtype. In contrast, smaller expansions (< 100 repeats), with fewer GGA interruptions and more AGC interruptions, are commonly seen in the movement disorder-dominant subtype[82,83]. Furthermore, repeat size has been shown to correlate negatively with age of onset in a cohort of 635 NIID patients[84].
HD
HD is an autosomal dominant neurodegenerative disease, classified according to age of onset into juvenile
HD is caused by an abnormal expansion of CAG trinucleotide repeats in exon 1 of the HTT gene on chromosome 4[86]. In unaffected individuals, the number of CAG repeats is ≤ 26. Carriers with 27-35 repeats do not develop HD, but the repeat length may expand when transmitted to offspring, increasing their risk of disease. Repeat lengths of 36~39 are associated with reduced penetrance, while ≥ 40 repeats confer full penetrance, and all carriers eventually develop HD[87]. It is important to distinguish HD from HD-like (HDL) diseases when HTT repeat lengths fall within the normal range. For example, HDL1, caused by PRNP mutations, and HDL2, resulting from abnormal JPH3 CTG repeat expansions, can exhibit clinical phenotypes that closely resemble HD[88,89].
PRION DISEASE
Prion diseases are a group of fatal, rapidly progressive neurodegenerative disorders caused by misfolded prion proteins. The primary pathological features include spongiform changes and gliosis in the central nervous system[90,91]. Human prion diseases primarily include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), fatal familial insomnia (FFI), and Kuru disease. CJD is the most common prion disease, characterized clinically by rapidly progressive dementia, myoclonus, and ataxia[92]. GSS typically presents with cerebellar ataxia and cognitive impairment, while FFI is primarily associated with progressive sleep disturbances and autonomic dysfunction. Prion diseases occur in sporadic, genetic, and acquired forms[93].
Genetic forms account for approximately 10%-15% of human prion diseases[94]. To date, more than 60 PRNP variants have been identified[95]. The PRNP gene, located on chromosome 2, encodes the 253 amino acid PRNP protein. Reported PRNP variants include missense mutations, octapeptide repeats insertions (OPRIs), octapeptide repeats deletions (OPRDs), and premature termination variants[96]. The frequency of specific variants varies significantly across populations. The E200K and V210I variants are the most common in Europe and the USA, with E200K particularly prevalent in Slovakia, Israel, Italy, and Chile[97,98]. Different PRNP variants are associated with distinct symptoms, age of onset, and disease duration. Some variants, such as OPRI, A117V, and G114V, are fully penetrant and typically manifest in early adulthood[99].
Approximately 85% of human prion disease cases are sporadic CJD (sCJD). The cause of sCJD remains largely unknown, aside from aging and certain genetic risk factors[100]. Among these, the PRNP M129V polymorphism is the strongest known genetic risk factor[101]. Specifically, in codon 129 of PRNP, about 70% of sCJD patients carry the MM genotype, 16% the VV genotype, and 13% the MV genotype[102]. GWASs have identified several additional genetic variants that influence sCJD risk, including PRNP rs1799990 (M129V)[103], PRNP rs6107516, and GRM8 rs6951643[104]. A recent large GWAS also revealed that STX6 rs3747957 and GAL3ST1 rs2267161 are associated with sCJD in a cohort of 5,208 cases[105]. Furthermore, gene-based analysis of 3,767 sCJD cases demonstrated a significant association between HS6ST3 and age at disease onset[106]. In contrast, sequencing of 205 sCJD cases and 170 controls found no pathogenic somatic PRNP mutations, suggesting that somatic mutations may not play a major role in sCJD[107].
CONCLUSION
In summary, genetic factors play a crucial role in neurodegenerative dementias, including both phenotypic expression and disease onset. In AD, more than 70 common loci have been implicated, alongside dozens of rare variants that contribute significantly to disease risk. In FTLD, variants in causal genes such as MAPT, C9orf72, and GRN are major contributors. DLB exhibits genetic overlap with both AD and PD. NIID is primarily associated with repeat expansions in the NOTCH2NLC gene, particularly within the Chinese population. HD results from abnormal CAG repeat expansions in the HTT gene. Although rare, prion diseases demonstrate strong genetic determinants, with PRNP variants influencing disease presentation. Collectively, these findings highlight the pivotal role of genetic mechanisms in neurodegenerative dementias and emphasize the need for further studies to elucidate their genetic basis.
DECLARATIONS
Authors’ contributions
Literature search, writing, and original draft preparation: Xiao X, Luo S, Li J
Conceptualization, review, revision, and editing: Shen L, Jiao B
Availability of data and materials
Not applicable.
Financial support and sponsorship
This study was supported by the National Natural Science Foundation of China (U22A20300, 82502244, 82371434, 82071216), STI2030-Major Projects (2021ZD0201803), National Key R&D Program of China (2023YFC3603700), Outstanding Youth Fund of Hunan Provincial Natural Science Foundation (2024JJ2097), Hunan Health Commission Grant (20232460), Science and Technology Major Project of Hunan Province (2021SK1020), Postdoctoral Fellowship Program of CPSF (No.GZC20233185), and the Scientific Research Program of FuRong Laboratory (No.2024PT5108).
Conflicts of interest
Lu Shen is an Editorial Board member of the journal Ageing and Neurodegenerative Diseases. She was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, or decision making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7:e105-25.
2. Mukadam N, Wolters FJ, Walsh S, et al. Changes in prevalence and incidence of dementia and risk factors for dementia: an analysis from cohort studies. Lancet Public Health. 2024;9:e443-60.
3. Koriath CAM, Kenny J, Ryan NS, et al. Genetic testing in dementia - utility and clinical strategies. Nat Rev Neurol. 2021;17:23-36.
4. Seath P, Macedo-Orrego LE, Velayudhan L. Clinical characteristics of early-onset versus late-onset Alzheimer’s disease: a systematic review and meta-analysis. Int Psychogeriatr. 2024;36:1093-109.
5. Sims R, Hill M, Williams J. The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci. 2020;23:311-22.
6. Xiao X, Liu H, Liu X, Zhang W, Zhang S, Jiao B.
7. Nudelman KNH, Jackson T, Rumbaugh M, et al; DIAN/DIAN-TU Clinical/Genetics Committee. Pathogenic variants in the Longitudinal Early-onset Alzheimer’s Disease Study cohort. Alzheimers Dement. 2023;19 Suppl 9:S64-73.
8. Jiao B, Liu H, Guo L, et al. The role of genetics in neurodegenerative dementia: a large cohort study in South China. NPJ Genom Med. 2021;6:69.
9. Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20:68-80.
10. Belloy ME, Andrews SJ, Le Guen Y, et al. APOE genotype and Alzheimer disease risk across age, sex, and population ancestry. JAMA Neurol. 2023;80:1284-94.
11. Fortea J, Pegueroles J, Alcolea D, et al. APOE4 homozygozity represents a distinct genetic form of Alzheimer’s disease. Nat Med. 2024;30:1284-91.
12. Chemparathy A, Le Guen Y, Chen S, et al. APOE loss-of-function variants: compatible with longevity and associated with resistance to Alzheimer’s disease pathology. Neuron. 2024;112:1110-1116.e5.
13. Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088-93.
14. Lambert JC, Heath S, Even G, et al; European Alzheimer’s Disease Initiative Investigators. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094-9.
15. Hollingworth P, Harold D, Sims R, et al; Alzheimer’s Disease Neuroimaging Initiative. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429-35.
16. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436-41.
17. Lambert JC, Ibrahim-Verbaas CA, Harold D, et al; European Alzheimer’s Disease Initiative (EADI). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452-8.
18. Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404-13.
19. Kunkle BW, Grenier-Boley B, Sims R, et al; Alzheimer Disease Genetics Consortium (ADGC). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414-30.
20. Wightman DP, Jansen IE, Savage JE, et al; 23andMe Research Team. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53:1276-82.
21. Bellenguez C, Küçükali F, Jansen IE, et al; EADB. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54:412-36.
22. Andrews SJ, Renton AE, Fulton-Howard B, Podlesny-Drabiniok A, Marcora E, Goate AM. The complex genetic architecture of Alzheimer’s disease: novel insights and future directions. EBioMedicine. 2023;90:104511.
23. Jiao B, Xiao X, Yuan Z, et al. Associations of risk genes with onset age and plasma biomarkers of Alzheimer’s disease: a large case-control study in mainland China. Neuropsychopharmacology. 2022;47:1121-7.
24. Zhou X, Chen Y, Mok KY, et al; Alzheimer’s Disease Neuroimaging Initiative. Identification of genetic risk factors in the Chinese population implicates a role of immune system in Alzheimer’s disease pathogenesis. Proc Natl Acad Sci U S A. 2018;115:1697-706.
25. Jia L, Li F, Wei C, et al. Prediction of Alzheimer’s disease using multi-variants from a Chinese genome-wide association study. Brain. 2021;144:924-37.
26. Ge YJ, Chen SD, Wu BS, et al. Genome-wide meta-analysis identifies ancestry-specific loci for Alzheimer’s disease. Alzheimers Dement. 2024;20:6243-56.
27. Guerreiro R, Wojtas A, Bras J, et al; Alzheimer Genetic Analysis Group.
28. Cruchaga C, Karch CM, Jin SC, et al; Alzheimer’s Research UK (ARUK) Consortium. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505:550-4.
29. Roeck A, Van Broeckhoven C, Sleegers K. The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019;138:201-20.
30. Wetzel-Smith MK, Hunkapiller J, Bhangale TR, et al; Alzheimer’s Disease Genetics Consortium. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat Med. 2014;20:1452-7.
31. Campion D, Charbonnier C, Nicolas G.
32. Sims R, van der Lee SJ, Naj AC, et al; ARUK Consortium. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49:1373-84.
33. Holstege H, Hulsman M, Charbonnier C, et al. Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer’s disease. Nat Genet. 2022;54:1786-94.
34. Qin W, Zhou A, Zuo X, et al. Exome sequencing revealed PDE11A as a novel candidate gene for early-onset Alzheimer’s disease. Hum Mol Genet. 2021;30:811-22.
35. Zhang DF, Fan Y, Xu M, et al; Alzheimer’s Disease Neuroimaging Initiative (ADNI).
36. Luo R, Fan Y, Yang J, et al. A novel missense variant in ACAA1 contributes to early-onset Alzheimer’s disease, impairs lysosomal function, and facilitates amyloid-β pathology and cognitive decline. Signal Transduct Target Ther. 2021;6:325.
37. Liao X, Cai F, Sun Z, et al. Identification of Alzheimer’s disease-associated rare coding variants in the ECE2 gene. JCI Insight. 2020;5:135119.
38. Jiang Y, Wan M, Xiao X, et al. GSN gene frameshift mutations in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2023;94:436-47.
39. Xiao X, Liu H, Yao R, et al.
40. Collins RL, Talkowski ME. Diversity and consequences of structural variation in the human genome. Nat Rev Genet. 2025;26:443-62.
41. Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16:172-83.
42. Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016;12:733-48.
43. Lee WP, Tucci AA, Conery M, et al. Copy number variation identification on 3,800 Alzheimer’s disease whole genome sequencing data from the Alzheimer’s disease sequencing project. Front Genet. 2021;12:752390.
44. Ming C, Wang M, Wang Q, et al. Whole genome sequencing-based copy number variations reveal novel pathways and targets in Alzheimer’s disease. Alzheimers Dement. 2022;18:1846-67.
46. Roeck A, Duchateau L, Van Dongen J, et al; BELNEU Consortium. An intronic VNTR affects splicing of ABCA7 and increases risk of Alzheimer’s disease. Acta Neuropathol. 2018;135:827-37.
47. Jiao B, Zhou L, Zhou Y, et al. Identification of expanded repeats in NOTCH2NLC in neurodegenerative dementias. Neurobiol Aging. 2020;89:142.e1-7.
48. Wu W, Yu J, Qian X, et al. Intermediate-length CGG repeat expansion in NOTCH2NLC is associated with pathologically confirmed Alzheimer’s disease. Neurobiol Aging. 2022;120:189-95.
49. Zhang X, Farrell JJ, Tong T, et al; Alzheimer’s Disease Sequencing Project. Association of mitochondrial variants and haplogroups identified by whole exome sequencing with Alzheimer’s disease. Alzheimers Dement. 2022;18:294-306.
50. Grossman M, Seeley WW, Boxer AL, et al. Frontotemporal lobar degeneration. Nat Rev Dis Primers. 2023;9:40.
51. Mol MO, van Rooij JGJ, Wong TH, et al. Underlying genetic variation in familial frontotemporal dementia: sequencing of 198 patients. Neurobiol Aging. 2021;97:148.e9-148.e16.
52. Leveille E, Ross OA, Gan-Or Z. Tau and MAPT genetics in tauopathies and synucleinopathies. Parkinsonism Relat Disord. 2021;90:142-54.
53. Amin S, Carling G, Gan L. New insights and therapeutic opportunities for progranulin-deficient frontotemporal dementia. Curr Opin Neurobiol. 2022;72:131-9.
54. Tang X, Toro A, T G S, et al. Divergence, convergence, and therapeutic implications: a cell biology perspective of C9ORF72-ALS/FTD. Mol Neurodegener. 2020;15:34.
55. Grover A, Houlden H, Baker M, et al. 5’ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274:15134-43.
56. Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24:277-95.
57. Woollacott IO, Mead S. The C9ORF72 expansion mutation: gene structure, phenotypic and diagnostic issues. Acta Neuropathol. 2014;127:319-32.
58. Majounie E, Renton AE, Mok K, et al; Chromosome 9-ALS/FTD Consortium. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323-30.
59. Xu T, Weng L, Zhang C, et al. Genetic spectrum features and diagnostic accuracy of four plasma biomarkers in 248 Chinese patients with frontotemporal dementia. Alzheimers Dement. 2024;20:7281-95.
60. Huang M, Modeste E, Dammer E, et al. Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathol Commun. 2020;8:163.
61. Jiao B, Xiao T, Hou L, et al. High prevalence of CHCHD10 mutation in patients with frontotemporal dementia from China. Brain. 2016;139:e21.
62. Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234-9.
63. Ferrari R, Hernandez DG, Nalls MA, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014;13:686-99.
64. Pottier C, Ren Y, Perkerson RB 3rd, et al. Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol. 2019;137:879-99.
65. Coppola G, Chinnathambi S, Lee JJ, et al; Alzheimer’s Disease Genetics Consortium. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet. 2012;21:3500-12.
66. Reus LM, Jansen IE, Mol MO, et al. Genome-wide association study of frontotemporal dementia identifies a C9ORF72 haplotype with a median of 12-G4C2 repeats that predisposes to pathological repeat expansions. Transl Psychiatry. 2021;11:451.
67. Kaivola K, Chia R, Ding J, et al; American Genome Center. Genome-wide structural variant analysis identifies risk loci for non-Alzheimer’s dementias. Cell Genom. 2023;3:100316.
68. Jellinger KA, Korczyn AD. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 2018;16:34.
69. Palushaj B, Lewis SJG, Abdelnour C. What is the future for dementia with Lewy bodies? J Neurol. 2024;272:43.
70. Guerreiro R, Ross OA, Kun-Rodrigues C, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol. 2018;17:64-74.
71. Chia R, Sabir MS, Bandres-Ciga S, et al; American Genome Center. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet. 2021;53:294-303.
72. Larsson V, Torisson G, Londos E. Relative survival in patients with dementia with Lewy bodies and Parkinson’s disease dementia. PLoS One. 2018;13:e0202044.
73. Guella I, Evans DM, Szu-Tu C, et al; SNCA Cognition Study Group. α-synuclein genetic variability: a biomarker for dementia in Parkinson disease. Ann Neurol. 2016;79:991-9.
74. Davis MY, Johnson CO, Leverenz JB, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. 2016;73:1217-24.
75. Winder-Rhodes SE, Hampshire A, Rowe JB, et al. Association between MAPT haplotype and memory function in patients with Parkinson’s disease and healthy aging individuals. Neurobiol Aging. 2015;36:1519-28.
76. Real R, Martinez-Carrasco A, Reynolds RH, et al. Association between the LRP1B and APOE loci and the development of Parkinson’s disease dementia. Brain. 2023;146:1873-87.
77. Tian Y, Zhou L, Gao J, et al. Clinical features of NOTCH2NLC-related neuronal intranuclear inclusion disease. J Neurol Neurosurg Psychiatry. 2022;93:1289-98.
78. Tai H, Wang A, Zhang Y, et al; China NIID Collaboration Alliance. Clinical features and classification of neuronal intranuclear inclusion disease. Neurol Genet. 2023;9:e200057.
79. Tian Y, Wang JL, Huang W, et al. Expansion of human-specific GGC repeat in neuronal intranuclear inclusion disease-related disorders. Am J Hum Genet. 2019;105:166-76.
80. Sone J, Mitsuhashi S, Fujita A, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet. 2019;51:1215-21.
81. Chen Z, Yan Yau W, Jaunmuktane Z, et al; Genomics England Research Consortium. Neuronal intranuclear inclusion disease is genetically heterogeneous. Ann Clin Transl Neurol. 2020;7:1716-25.
82. Huang XR, Tang BS, Jin P, Guo JF. The phenotypes and mechanisms of NOTCH2NLC-related GGC repeat expansion disorders: a comprehensive review. Mol Neurobiol. 2022;59:523-34.
83. Chen Z, Xu Z, Cheng Q, et al. Phenotypic bases of NOTCH2NLC GGC expansion positive neuronal intranuclear inclusion disease in a Southeast Asian cohort. Clin Genet. 2020;98:274-81.
84. Zeng T, Chen Y, Huang H, et al. Neuronal intranuclear inclusion disease with NOTCH2NLC GGC repeat expansion: a systematic review and challenges of phenotypic characterization. Aging Dis. 2024;16:578-97.
87. Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204-16.
88. Krause A, Anderson DG, Ferreira-Correia A, et al. Huntington disease-like 2: insight into neurodegeneration from an African disease. Nat Rev Neurol. 2024;20:36-49.
90. Zerr I, Ladogana A, Mead S, Hermann P, Forloni G, Appleby BS. Creutzfeldt-Jakob disease and other prion diseases. Nat Rev Dis Primers. 2024;10:14.
91. Baiardi S, Rossi M, Capellari S, Parchi P. Recent advances in the histo-molecular pathology of human prion disease. Brain Pathol. 2019;29:278-300.
92. Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis. 2020;20:e2-e10.
93. Piñar-Morales R, Barrero-Hernández F, Aliaga-Martínez L. Human prion diseases: an overview. Med Clin. 2023;160:554-60.
94. Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64:1586-91.
95. Mead S, Lloyd S, Collinge J. Genetic factors in mammalian prion diseases. Annu Rev Genet. 2019;53:117-47.
96. Kim MO, Takada LT, Wong K, Forner SA, Geschwind MD. Genetic PrP prion diseases. Cold Spring Harb Perspect Biol. 2018;10:a033134.
97. Lee HS, Sambuughin N, Cervenakova L, et al. Ancestral origins and worldwide distribution of the PRNP 200K mutation causing familial Creutzfeldt-Jakob disease. Am J Hum Genet. 1999;64:1063-70.
98. Brown P, Gálvez S, Goldfarb LG, et al. Familial Creutzfeldt-Jakob disease in Chile is associated with the codon 200 mutation of the PRNP amyloid precursor gene on chromosome 20. J Neurol Sci. 1992;112:65-7.
99. Mead S, Poulter M, Beck J, et al. Inherited prion disease with six octapeptide repeat insertional mutation--molecular analysis of phenotypic heterogeneity. Brain. 2006;129:2297-317.
100. McDonough GA, Cheng Y, Morillo K, et al. Neuropathologically-directed profiling of PRNP somatic and germline variants in sporadic human prion disease. bioRxiv. ;2024:2024.
101. Kim YC, Jeong BH. The first meta-analysis of the M129V single-nucleotide polymorphism (SNP) of the prion protein gene (PRNP) with sporadic Creutzfeldt-Jakob disease. Cells. 2021;10:3132.
102. Alperovitch A, Zerr I, Pocchiari M, et al. Codon 129 prion protein genotype and sporadic Creutzfeldt-Jakob disease. Lancet. 1999;353:1673-4.
103. Mead S, Uphill J, Beck J, et al. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum Mol Genet. 2012;21:1897-906.
104. Sanchez-Juan P, Bishop MT, Kovacs GG, et al. A genome wide association study links glutamate receptor pathway to sporadic Creutzfeldt-Jakob disease risk. PLoS One. 2014;10:e0123654.
105. Jones E, Hummerich H, Viré E, et al. Identification of novel risk loci and causal insights for sporadic Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol. 2020;19:840-8.
106. Hummerich H, Speedy H, Campbell T, et al. Genome wide association study of clinical duration and age at onset of sporadic CJD. PLoS One. 2024;19:e0304528.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.