

AI agents for solid electrolytes: opportunities, challenges, and future directions
 0
0 
Abstract
Artificial intelligence (AI) and autonomous agents are transforming the discovery and optimization of solid electrolytes, a class of materials crucial to the safety and performance of next-generation batteries. This review summarizes recent progress in integrating machine learning, molecular dynamics, and density functional theory within closed-loop or semi-autonomous workflows that accelerate the evaluation of ionic conductivity, electrochemical and chemical stability, and processability. Data-driven frameworks now accelerate the screening of sulfides, oxides, and halides, while phase-field and multiscale models have provided mechanistic insight into dendrite formation, interfacial degradation, and chemo-mechanical coupling. Autonomous laboratories that combine robotic synthesis, in situ characterization, and Bayesian optimization further enable closed-loop experimental discovery. Despite this progress, challenges remain in data quality, model interpretability, and the limited autonomy of current systems. Future development will rely on five key directions: (1) constructing interoperable multiscale databases, (2) developing explainable and data-efficient algorithms, (3) tightly integrating computation with experiment, (4) exploring new solid-electrolyte chemistries via agent-driven optimization, and (5) fostering coordinated global collaboration among open AI agents. Together, these developments mark a transition from empirical discovery to an integrated, self-improving research paradigm, where AI evolves from a predictive assistant into an active collaborator that learns, reasons, and supports materials innovation alongside human researchers.
Keywords
INTRODUCTION
Energy storage has evolved into a global research priority, with governments and industry investing heavily to achieve net-zero targets[1]. Electric vehicles, as a cornerstone of low-carbon mobility, are expanding rapidly, intensifying the demand for advanced batteries[2]. Commercial lithium-ion batteries (LIBs) rely on liquid organic electrolytes, which present risks such as leakage, flammability, and dendrite formation[3]. Solid electrolytes (SEs) have therefore emerged as a promising alternative, offering non-flammability, improved mechanical robustness, and compatibility with high-energy-density electrodes[4,5]. An ideal SE should simultaneously combine high ionic conductivity [> 10-3 S cm-1 at room temperature (RT)][6], negligible electronic conductivity, a wide electrochemical stability window (ESW, > 4 V)[7], and stable electrode-electrolyte interfaces[8]. Achieving high conductivity, wide electrochemical stability, low electronic leakage, and durable solid-solid interfaces remains challenging[9]. To overcome these intertwined material and performance challenges, the evolution of scientific discovery itself has become a key enabler of progress.
The trajectory of scientific discovery has evolved from empirical observation and trial-and-error experimentation to theory-driven exploration grounded in physical and chemical principles[10] and later to computational modeling enabled by methods such as density functional theory (DFT), molecular dynamics (MD), and Monte Carlo simulations[11] [Figure 1A]. In recent years, data-driven science has further accelerated this process through the integration of machine learning (ML), materials informatics, and high-throughput computational and experimental techniques[17]. Despite these advances, current computational approaches remain resource-intensive and face challenges in multiscale modeling, particularly in bridging electrochemical interfaces and structure-property relationships. These limitations highlight the need for new research frameworks built on artificial intelligence (AI)[18]. Advances in ML, natural language processing (NLP)[19,20], and optimization algorithms are increasingly regarded as laying the foundation of a new era of scientific discovery driven by advanced AI agents [Figure 1A][21].
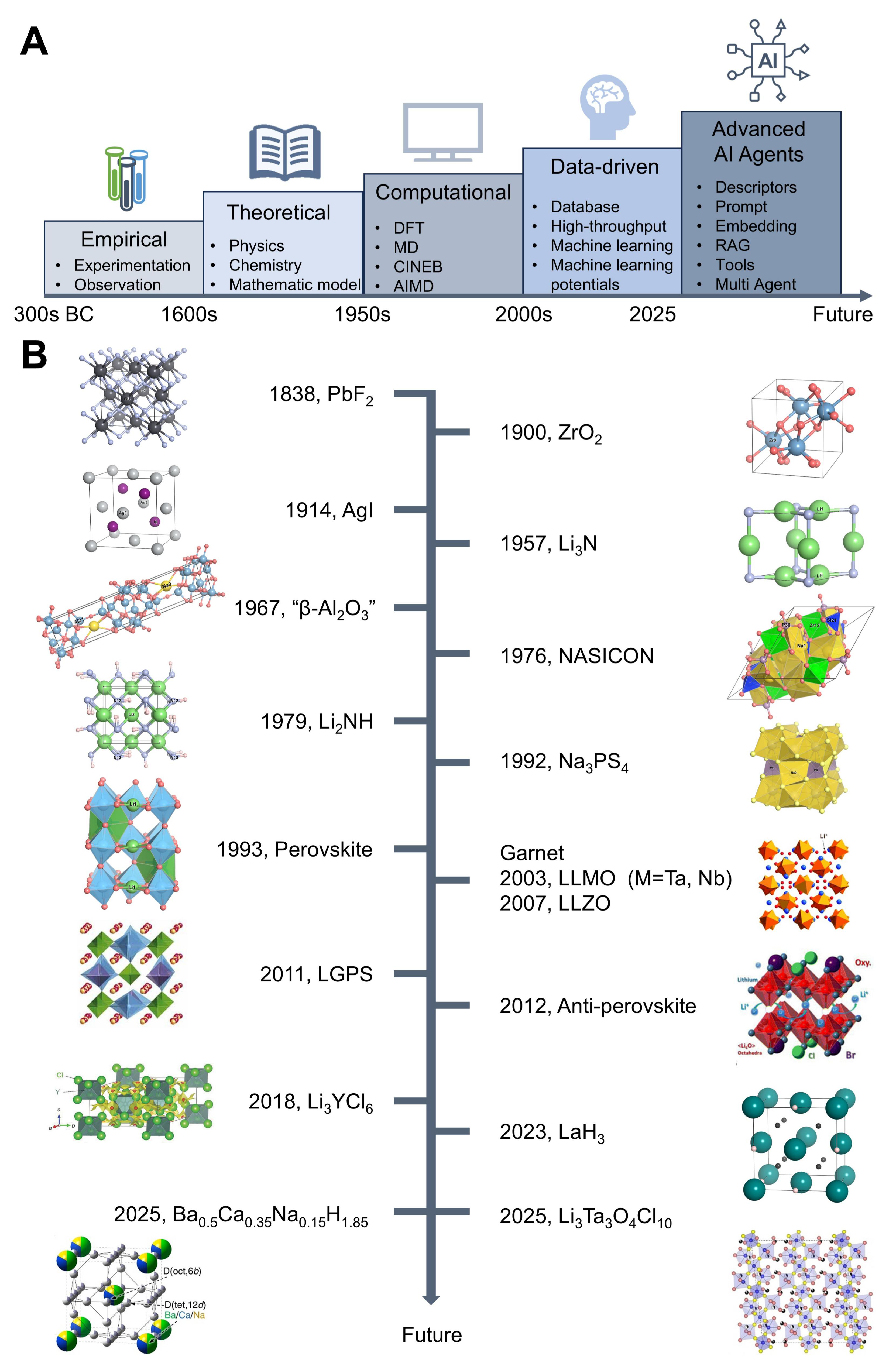
Figure 1. (A) Evolution of scientific discovery paradigms and (B) Historical development of SEs, reproduced with permission from Refs.[6,12-16]. Copyright 2011, Springer Nature; 2007, 2018, Wiley-VCH; 2012, American Chemical Society; 2025, AAAS. DFT: Density functional theory; MD: molecular dynamics; CINEB: climbing image nudged elastic band; AIMD: ab initio molecular dynamics; AI: artificial intelligence; RAG: retrieval-augmented generation; NASICON: sodium superionic conductor; LLTO: Li3xLa2/3-xTiO3; LLZO: Li7La3Zr2O12; LLMO: Li5La3M2O12; LGPS: Li10GeP2S12; SE: solid electrolyte.
Historically, research on SE relied on trial-and-error synthesis and experimental validation, which is slow and resource-intensive[22]. Advanced computational techniques have become indispensable for probing features that are difficult to access experimentally, such as ion-conduction pathways and correlated transport mechanisms[23-25]. From the 2000s onward, high-throughput platforms and large databases [e.g., Materials Project (MP) and Inorganic Crystal Structure Database (ICSD)] enabled large-scale data accumulation. Since the mid-2010s, ML methods have leveraged these datasets to predict ionic conductivity, stability windows, and lattice dynamics, screening thousands of candidates in a fraction of the time and enabling rapid virtual screening of candidate materials[26-30]. However, these approaches remain primarily predictive. They can reveal correlations and rank materials, but cannot autonomously design synthesis strategies or adaptively guide experiments. Recent progress is therefore shifting from predictive models to autonomous AI agents capable of reasoning, planning, and adapting within closed discovery loops, thereby accelerating the path from design to realization. This methodological shift mirrors the acceleration of materials innovation in Figure 1B, where rapid progress in sulfides, garnets, and halides coincides with the rise of database-driven screening and ML-enabled design.
Figure 1B summarizes key milestones in SE development, which progressed in parallel with the evolution of discovery paradigms in Figure 1A, from empirical trial-and-error to theory- and data-driven design. In the early nineteenth century, Faraday demonstrated ionic conduction in Ag2S and PbF2, establishing the foundation of ion transport in solids[31]. By the early twentieth century, Nernst revealed ionic conduction in doped zirconia and formulated the Nernst equation, thereby laying the foundation for modern defect chemistry[32]. The discovery of the superionic phase α-AgI by Tubandt and Lorenz in the 1910s[33] and the development of point-defect theory by Frenkel, Schottky, and Wagner in the 1930s established the conceptual basis of fast-ion conduction[34]. The modern era began in the 1960s with the development of β-alumina for Na-S batteries by Yao and Kummer[35], followed by Takahashi’s reports on Ag3SI and Rb4Cu16I7Cl13, which exhibited conductivities approaching 0.3 S cm-1 at RT[36]. In 1976, Goodenough and Hong introduced the NASICON family (Na1+xZr2P3-xSixO12)[37], and in 1980, Hong proposed its lithium analog [(lithium superionic conductor) LISICON], establishing structural design principles that continue to guide SE development[38]. Hydride conductors were first recognized as SEs in 1979 with the discovery of Li2NH[39].
In 1992, Na3PS4 was first investigated as a sulfide SE, exhibiting an ionic conductivity of 2 × 10-4 S cm-1 at RT[40]. In 2000, Kanno et al. reported the thio-LISICON compound Li3.25Ge0.25P0.75S4, which exhibited an ionic conductivity of 6.5 × 10-5 S cm-1 at RT among sulfide electrolytes[41]. A breakthrough came in 2011 with Li10GeP2S12 (LGPS), a superionic conductor with an ionic conductivity of 1.2 × 10-2 S cm-1 at RT and rapidly accelerated research on sulfide SEs[6]. In parallel, oxide garnet electrolytes were explored. In 1993, Inaguma et al.[42] reported perovskite-type SEs, Li3xLa2/3-xTiO3 (LLTO), which exhibited a total ionic conductivity exceeding 2 × 10-6 S cm-1 at RT but were incompatible with Li (stable only above ~ 1.8 V)[42,43]. In 2003, Thangadurai et al. synthesized Li5La3M2O12 (LLMO, M = Ta, Nb), which exhibited an ionic conductivity of 10-6 S cm-1 at RT and provided stable interfaces with molten Li[44]. In 2007, Li7La3Zr2O12 (LLZO) was reported to exhibit ionic conductivity above 10-4 S cm-1 at RT and high stability[12]. In 2012, Li-rich anti-perovskites such as Li3OCl0.5Br0.5 were reported, exhibiting a RT ionic conductivity of 1.94 × 10-3 S cm-1[13]. In 2018, Asano et al. developed the trigonal Li3YCl6 and monoclinic Li3YBr6, which exhibited RT ionic conductivities of 5.1 × 10-4 and 1.7 × 10-3 S cm-1, respectively[14]. H- conduction in bulk materials was first reported by Verbraeken et al.[45,46] and verified by Kobayashi et al.[47]. Later, Takeiri et al. reported H- superionic conductivity of Ba-Li oxyhydride[48]. In 2023, Zhang et al. identified LaH3 as an H- ion conductor with an ionic conductivity of 1.0 × 10-2 S cm-1 at -40 °C[49] and further designed a core-shell hydride 3CeH3@BaH2, exhibiting an ionic conductivity of 10-3 S cm-1 at 60 °C and an initial battery capacity of 984 mAh g-1[50]. In the same year, Hirose et al. investigated an anti-α-AgI-type Ba0.5Ca0.35Na0.15H1.85 hydride SE that enabled a reversible capacity of 2,030 mAh g-1 in a Mg-H2 battery[49]. The most recent advance is Li3Ta3O4Cl10, a solid-state lithium tantalum oxychloride exhibiting an ionic conductivity of 1.37 × 10-2 S cm-1 at RT, attributed to lithium hopping among tetrahedral sites[15].
Together, these milestones chart the technological evolution of SEs and illustrate a gradual shift from empirical discovery to rational design. As the timeline advances, the coupled design space spanning composition, processing, and interfaces increasingly exceeds what empirical iteration can explore efficiently. In parallel, methodological progress has moved from theory and computation to data-driven screening and, more recently, toward agentic systems that integrate perception, reasoning, and action in closed-loop discovery. This convergence motivates the introduction of AI agents as a unifying framework for SE research. The two trajectories [Figure 1A and B] are now converging, as methodological innovation becomes increasingly aligned with technological progress. With the growing influence of AI, computational modeling, high-throughput data generation, and autonomous experimentation are being integrated into unified discovery frameworks. This convergence marks the beginning of a new stage in materials research, where AI agents are expected to transform the design and realization of SEs[51-53].
In this review, we first examine the contributions of computational and data-driven methods such as DFT, MD, and ML to SE research. We then discuss how AI agents extend these approaches toward autonomous adaptive discovery by integrating perception, reasoning, and action. Finally, the key challenges and future opportunities for developing agent-based frameworks that can accelerate the rational design and large-scale deployment of SEs are outlined.
DEFINITION AND SCOPE OF AI AGENTS IN SE DISCOVERY
AI agents differ fundamentally from traditional machine-learning models. Conventional ML models typically perform a single prediction task based on static training data, whereas an AI agent is an autonomous or semi-autonomous computational entity capable of perceiving information, reasoning about possible actions, executing tasks, and learning from feedback. In the context of SE research, AI agents serve as an overarching framework that integrates multi-step computational and experimental workflows (including data extraction, structure generation, simulation, optimization, and decision-making) into a coherent closed-loop discovery system.
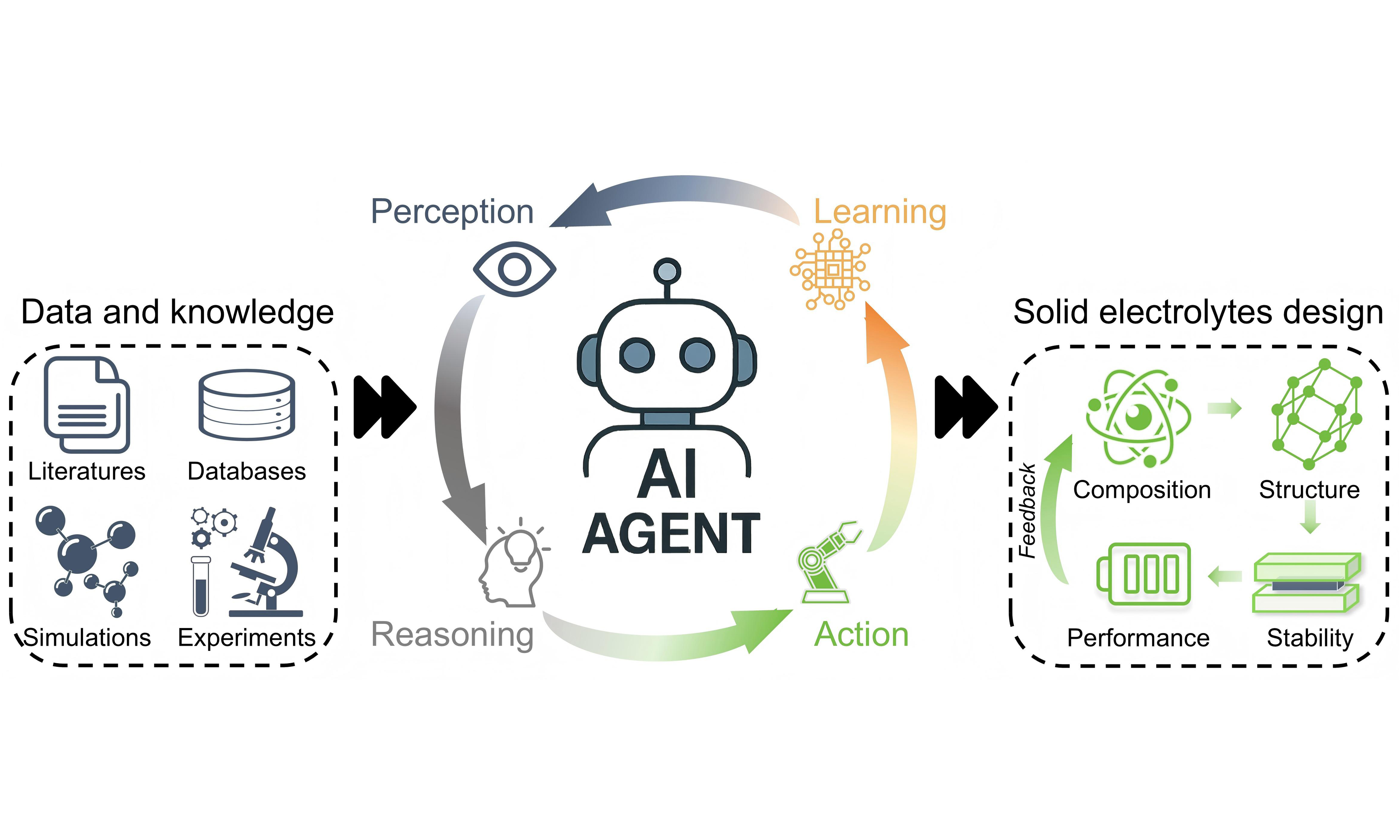
AI agents are generally characterized by four core capabilities [Figure 2]:
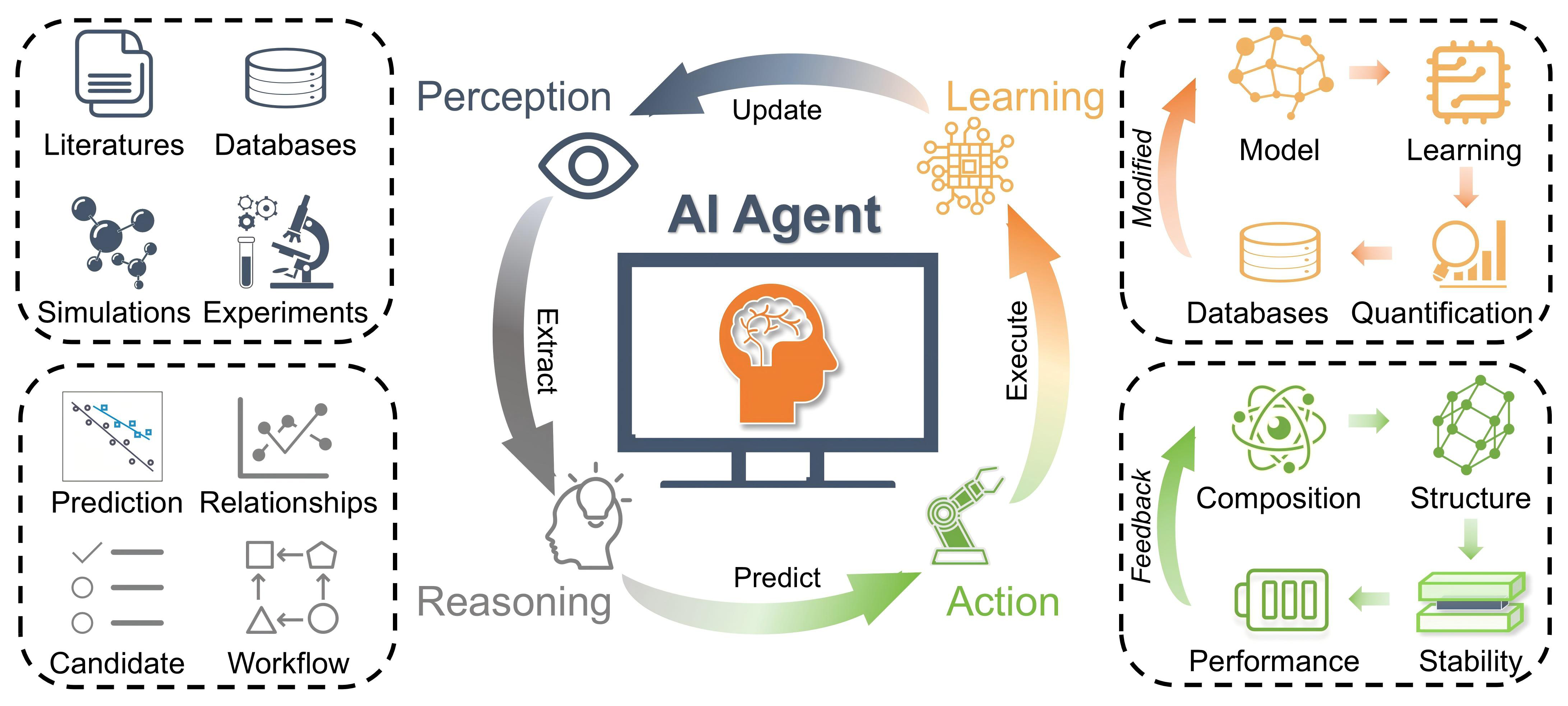
Figure 2. Schematic of an AI agent workflow in SE discovery. Perception extracts information, reasoning generates decisions, action executes tasks, and learning updates the agent, forming a closed-loop discovery system. AI: Artificial intelligence; SE: solid electrolyte.
Perception: the ability to ingest and interpret heterogeneous data sources, such as literature text, materials databases, DFT/ab initio molecular dynamics (AIMD) outputs (e.g., migration barriers, ESW, thermodynamic stability), and experimental measurements.
Reasoning: the ability to infer structure-property relationships, evaluate hypotheses, prioritize candidate SE materials, and plan multi-step workflows based on predicted ionic transport or electrochemical stability.
Action: the capability to perform operations such as querying materials databases, executing DFT/AIMD simulations, generating new SE structures, or triggering automated or human-in-the-loop experimental procedures.
Learning: the continuous refinement of internal models or policies based on newly acquired data, simulation feedback, failed predictions, or reinforcement signals, enabling adaptive improvement of the discovery process.
These capabilities collectively enable an AI agent to operate in a closed-loop manner: perception extracts information, reasoning generates decisions, action executes tasks, and learning updates the agent, thereby supporting autonomous and iterative SE discovery. Moreover, these capabilities distinguish AI agents from conventional ML predictors and justify a two-step narrative in this review: theoretical design principles that define what should be optimized are established in Section III, followed by computational, data-driven, and agent-enabled workflows that operationalize these objectives in later sections.
THEORETICAL DESIGN PRINCIPLES FOR SE
Building on the historical and methodological context discussed above, this section focuses on how theoretical design establishes the fundamental principles that guide modern SE development. The historical progression in Figure 1B shows that each new electrolyte family expands the performance envelope while simultaneously introducing distinct constraints in electrochemical stability, interfacial behavior, and manufacturability, making principled design criteria essential before any data-driven or agent-based optimization. Achieving high ionic conductivity while preserving chemical, electrochemical, and interfacial stability remains the central challenge in the rational design of these materials. Different SEs display intrinsic trade-offs among conductivity, electrochemical stability, and processability, reflecting the difficulty of satisfying all key performance requirements simultaneously.
Oxide electrolytes, such as garnet-type LLZO, exhibit excellent chemical stability and air tolerance but have relatively low RT ionic conductivities due to their rigid lattice[54]. Sulfide electrolytes such as LGPS exhibit liquid-like conductivities above 10-2 S cm-1[6]; however, their extreme moisture sensitivity generates toxic H2S and complicates large-scale processing[55]. Their high interfacial reactivity with cathodes further necessitates protective coatings to ensure electrochemical compatibility[56]. Halide SEs, represented by compounds such as Li3YCl6 and Li3YBr6, exhibit high ionic conductivities comparable to those of sulfide SEs and have therefore attracted increasing attention[14]. Their soft, polarizable anion frameworks enable fast Li+ transport, while their sensitivity to moisture and the cost of certain halide precursors remain practical limitations[57]. Complex hydrides achieve ionic conductivities near 10-3 S cm-1, but their narrow ESWs and moisture sensitivity restrict practical operation[58]. Polymer electrolytes, especially those based on poly(ethylene oxide) (PEO), provide mechanical flexibility and ease of processing, yet strong Li+ coordination with polymer chains limits their RT ionic conductivity[59,60]. These representative systems illustrate the inherent trade-offs between ionic conductivity, stability, and compatibility, explaining why empirical optimization alone has yielded only incremental progress.
Experimental discovery of SEs is further constrained by the vast chemical and structural design space. Exhaustively exploring compositional substitutions, dopants, and structural motifs through synthesis and testing would be prohibitively costly and time-consuming. Even high-throughput platforms, though more systematic, require significant resources and still struggle to capture the atomic-scale mechanisms governing ion migration and interfacial degradation[61-63]. For instance, aliovalent doping in garnet electrolytes improves Li+ conductivity, yet the microscopic effects of lattice distortion and defect chemistry remain difficult to isolate experimentally. Likewise, interfacial reactions at sulfide-oxide junctions involve charge-transfer and bond-reconstruction processes that are challenging to probe directly.
Theoretical design provides indispensable guidance for navigating this complexity. Methods rooted in quantum mechanics and atomistic simulations enable the prediction of thermodynamic stability, electronic structure, Li+ migration pathways, mechanical properties, and interfacial energetics before synthesis. By supplying such descriptors, theoretical methods narrow the search space and establish fundamental design rules. For example, correlating bottleneck size in oxide lattices with Li+ mobility or linking electronic structure to interfacial redox stability. These insights transform theoretical design into a predictive framework that complements experimental discovery and underpins data-driven and autonomous approaches to SE development.
EVOLUTION TOWARD AI-DRIVEN DISCOVERY
Quantum-level simulations
DFT simulations have long served as the foundation of atomistic modeling, enabling the investigation of intrinsic properties such as lattice and electronic band structures, ion migration barriers, thermodynamic stability, and interfacial energetics relevant to battery performance [Figure 3A]. Using the climbing-image nudged elastic band (CI-NEB) method, researchers have mapped Li+ diffusion pathways and quantified migration barriers in superionic conductors. For instance, simulations of Li1.3Al0.3Ti1.7(PO4)3 (LATP) revealed migration barriers of approximately 0.20 eV for multi-ion cooperative motion and 0.49 eV for isolated hopping. The lower migration barrier agrees well with experiments [Figure 3B][64]. While CI-NEB effectively identifies migration pathways, it treats ion motion as a static process at 0 K and neglects temperature and lattice dynamics. These simplifications limit its ability to capture the collective hopping and disorder effects that govern ion transport in SE. Recently, ML has revealed the spatial correlations underlying collective ion hopping based on AIMD datasets, demonstrating the potential of ML approaches for analyzing SEs[67]. However, a physical framework is still required to fully understand this phenomenon and to design appropriate descriptors. In this context, a mathematical model based on a directed graph representation successfully captures the temporal and spatial correlations of collective hopping and reproduces ionic conductivity as a proof of concept[65].
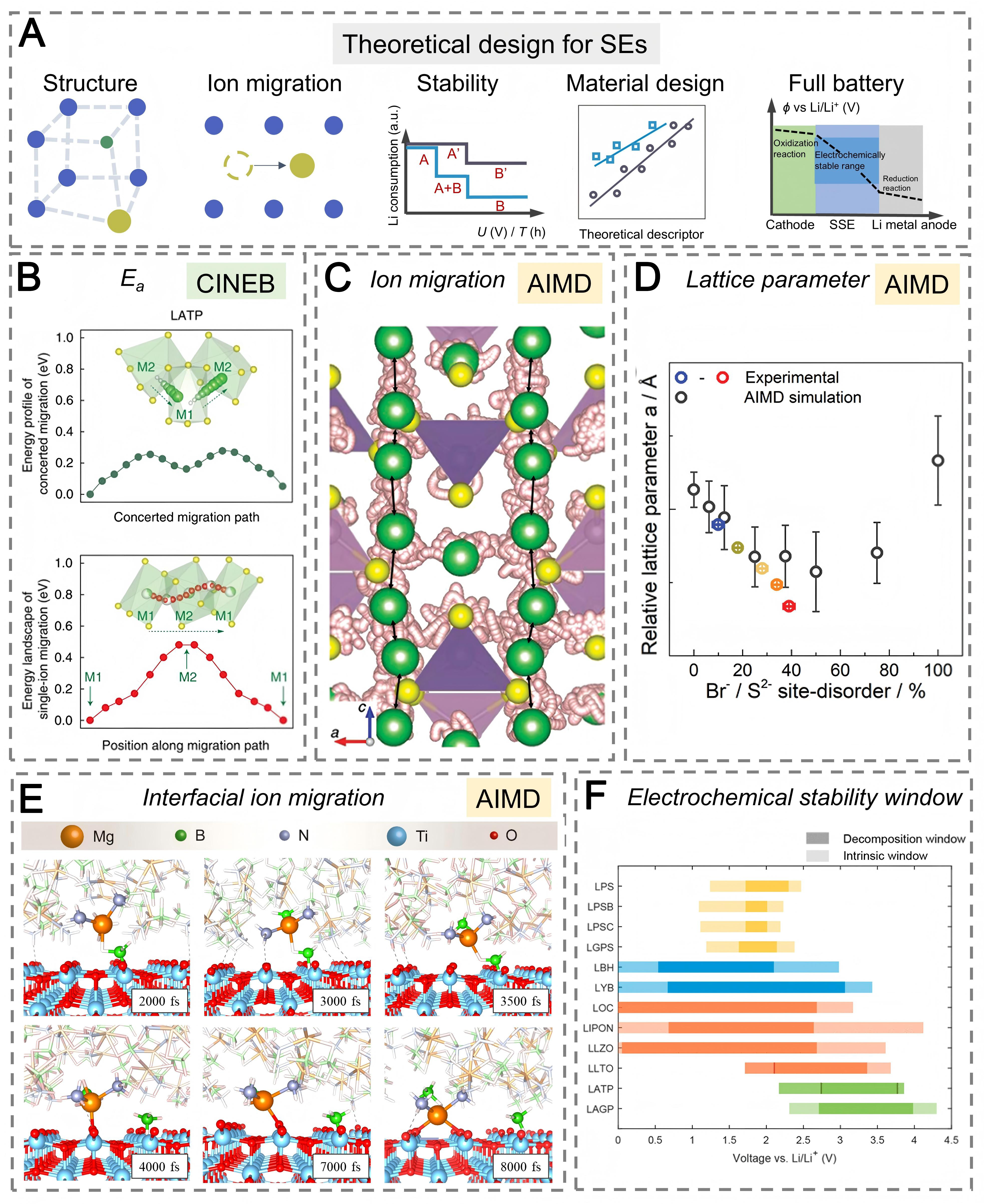
Figure 3. Computational simulation approaches for SEs. (A) Overview of theoretical techniques and representative predictive performance; (B) CI-NEB energy profiles showing Li+ migration pathway in LATP. Reproduced with permission from ref.[64]. Copyright 2017, Springer Nature; (C) AIMD trajectories of Li atoms (white) in LGPS, highlighting the one-dimensional c-axis diffusion. Reproduced with permission from ref.[26]. Copyright 2012, American Chemical Society; (D) Comparison between AIMD-predicted and experimentally measured lattice parameters of Li6PS5Br. Reproduced with permission from ref.[65]. Copyright 2021, Wiley-VCH; (E) Snapshots from AIMD simulations showing Mg2+ migration at the TiO2 (001) and Mg(BH4)2·1.5NH3 interface. Reproduced with permission from ref.[58]. Copyright 2022, Elsevier; (F) Calculated intrinsic and decomposition ESW of representative SEs. Reproduced with permission from ref.[66]. Copyright 2021, American Chemical Society. SE: Solid electrolyte; AIMD: ab initio molecular dynamics; CI-NEB: climbing-image nudged elastic band; LATP: Li1.3Al0.3Ti1.7(PO4)3; LGPS: Li10GeP2S12; ESW: electrochemical stability window.
AIMD combines first-principles accuracy with explicit time evolution, enabling the direct simulation of ion trajectories and diffusion coefficients at finite temperatures. Early studies established benchmarks for Li+ diffusivity and activation energies, providing mechanistic insight into superionic conduction. For instance, Mo et al. employed AIMD to reveal strong directional anisotropy in LGPS diffusion pathways [Figure 3C][26]. Xu et al. reproduced experimental ionic conductivities of Li6PS5Cl and Li6.25PS4O1.25Cl0.75 above RT with excellent quantitative agreement[69]. AIMD simulations have revealed that anion disorder governs Li+ transport in Li argyrodites [Figure 3D]: Specifically, substitutional S2-/X- disorder in Li6PS5Cl and Li6PS5I connects isolated diffusion channels into a three-dimensional Li+ network, whereas ordered anion frameworks confine Li+ motion and suppress macroscopic conductivity[65,70]. AIMD also enables mechanistic interrogation of interfacial ion transport, where simulations capture the progressive structural disordering at Li/Li6PS5Cl contacts and the corresponding evolution of Li+ migration barriers over time. The dynamic interfacial processes, including vacancy formation in Li metal and the densification of reaction layers on the electrolyte side, collectively regulate cross-interface transport and determine the magnitude of interfacial resistance[71]. Beyond Li-based systems, AIMD has also proven effective in probing sluggish multivalent-ion transport. In the hydride Mg(BH4)2·1.5NH3, AIMD simulations revealed a “coordination-unlock” mechanism at oxygen-deficient interfaces: oxygen vacancies destabilize Mg coordination shells and promote Mg2+ transport, yielding ionic conductivity near 10-4 S cm-1 at RT [Figure 3E][58].
Classical MD, employing empirical or developed machine-learned force fields, complements AIMD by extending timescales and system sizes. In sulfide SEs, both AIMD and classical MD simulations have demonstrated that anion-site disorder strongly correlates with enhanced Li+ conductivity[65,70]. In halide SEs, MD studies have revealed that rotational dynamics of the anion sublattice couple with Li+ migration, suggesting that a flexible lattice framework facilitates ionic transport[72]. In solid polymer electrolytes (SPEs), MD analysis showed the diffusion capacity of polymer chain segments on different fillers[73]. Methodological advances have further improved the efficiency of MD simulations. For example, a short-time correlation function approach originally developed for liquid electrolytes has been adapted to solid systems, reducing computational cost while maintaining accuracy[74]. Classical MD is not a quantum-level approach, as it relies on predefined empirical or machine-learned force fields rather than explicit quantum-mechanical calculations.
Nevertheless, AIMD remains computationally demanding, whereas classical MD and ML-based potentials rely on approximate interatomic potentials, which limit their scalability for large-scale compositional screening. These constraints have motivated the adoption of data-driven ML frameworks capable of reproducing first-principles accuracy at a fraction of the computational expense.
In addition to ion transport and interfacial energetics, DFT has also become essential for evaluating the ESW, a key property determining the compatibility of SEs with electrodes. Two thermodynamic formalisms are commonly used to quantify the ESW [Figure 3F][66]. The decomposition window is derived from the formation energies of the most favorable decomposition products at a given electrochemical potential using Li grand potential phase diagrams, with the nearest reduction and oxidation potentials defining the thermodynamic limits. However, this approach does not account for kinetic barriers and may underestimate the practical stability. The intrinsic window is obtained by calculating the energy changes associated with Li insertion and extraction within the SE lattice. These (de)lithiated phases often possess higher formation energies and may be metastable, representing indirect decomposition routes that become relevant once the SE reaches sufficiently unstable lithium compositions.
While both methods provide valuable thermodynamic descriptors, they do not capture decomposition pathways, reaction barriers, or kinetic stabilization at interfaces, all of which strongly influence the experimentally measured ESW. These limitations highlight the need for more integrated approaches that couple thermodynamics with kinetic considerations and thus motivate the development of ML-based frameworks discussed in Section Machine learning frameworks for SE discovery.
Machine learning frameworks for SE discovery
Data-driven methods have transformed SE discovery from intuition-driven trial-and-error into an iterative “learn-predict-verify” paradigm. Early supervised models such as logistic regression and support-vector machines demonstrated that even small experimental datasets could identify promising Li- and Mg-based conductors with significantly higher ionic conductivity than the mean population. These proof-of-concept studies established the feasibility of statistical screening and feature-based optimization. Subsequent advances introduced physics-informed descriptors [e.g., t/η (tolerance factor / atomic packing factor) ratios, phonon-derived features] that bridged empirical learning with physical interpretability, enabling targeted synthesis of high-conductivity antiperovskites and argyrodites.
Similar to classical force fields, machine-learning interatomic potentials (MLIPs) approximate the potential-energy surface without solving quantum-mechanical equations, although they achieve higher fidelity by training on ab initio data. Building upon these foundations, MLIPs now embed neural networks directly into the potential-energy formulation, achieving near-DFT accuracy for energies and forces while extending accessible time and length scales by several orders of magnitude. Universal MLIPs (uMLIPs) such as crystal hamiltonian graph neural network (GNN) (CHGNet), materials GNN with 3-body interactions (M3GNet), and message passing atomic cluster expansion (MACE) reproduce ionic-migration barriers and reveal structure-transport relationships across sulfide, oxide, and halide families. Integrating uncertainty quantification and fine-tuning into these frameworks paves the way for adaptive, agent-driven workflows.
ML for SE discovery
ML offers a scalable paradigm for property prediction across vast compositional spaces, serving as a data-efficient complement to ab initio modeling. Early supervised learning efforts demonstrated the feasibility of data-driven screening for SE discovery. Sendek et al. trained a logistic regression classifier on experimental conductivity data, narrowing 12,831 Li-containing compounds to 21 promising candidates [Figure 4A][27]. Fujimura et al. used a support vector regression (SVR) model to predict ionic conductivity in LISICON-type SEs, incorporating descriptors such as Li-ion diffusion coefficients, phase transition temperatures, and disordered structure volumes [Figure 4B][75]. These studies established a proof of concept that simple models trained on modest datasets meaningfully guide experimental prioritization and de-risk costly exploratory synthesis.
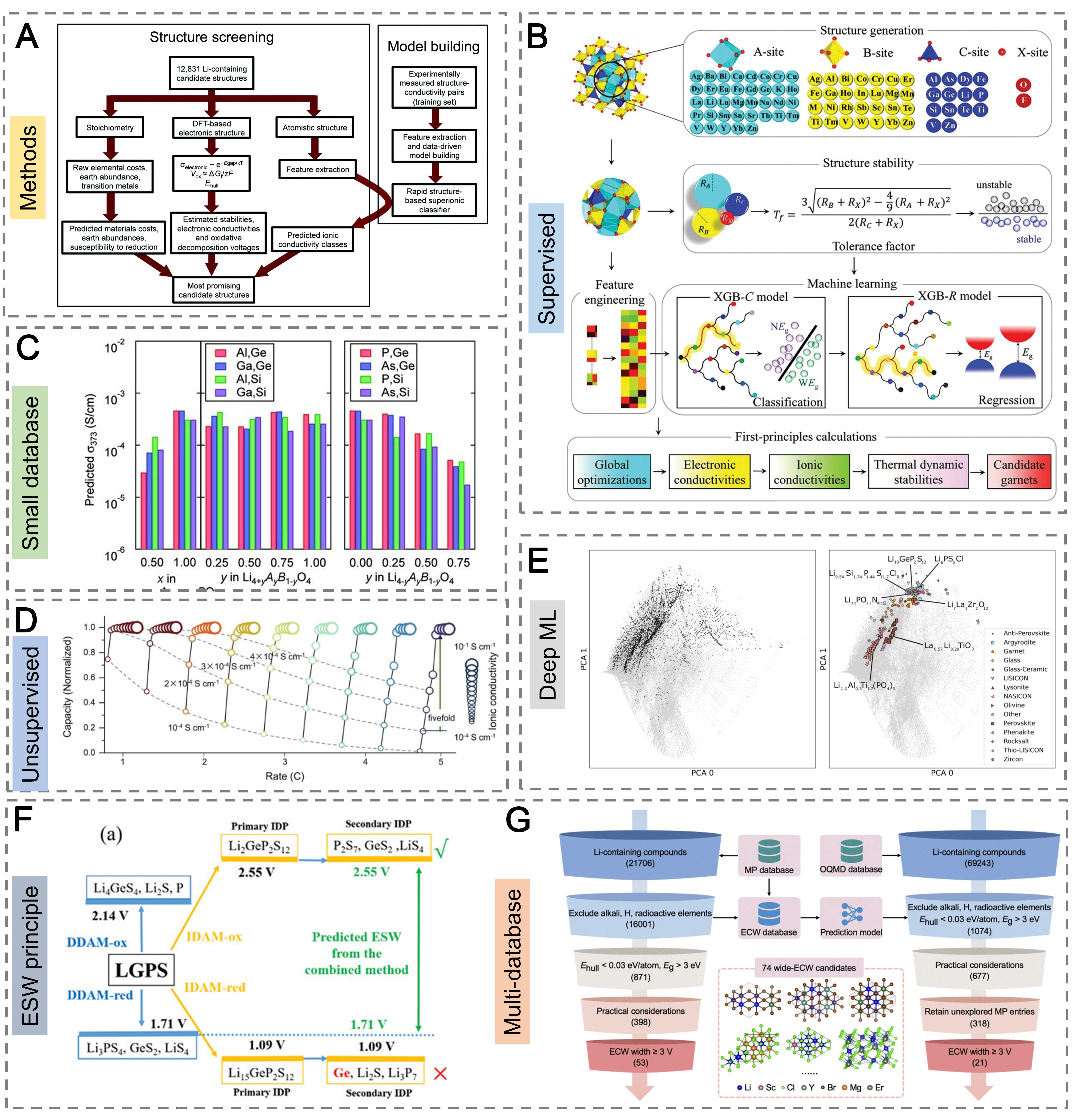
Figure 4. Evolution of ML applications in SE discovery. (A) Logistic regression screening of 12,831 Li-containing compounds using ML. Reproduced with permission from ref.[27]. Copyright 2017, Royal Society of Chemistry; (B) SVR prediction of ionic conductivity in LISICON-type electrolytes using a small database. Reproduced with permission from ref.[75]. Copyright 2013, Wiley-VCH; (C) ML framework accelerated garnet-based SE screening supervised by the MP database. Reproduced with permission from ref.[76]. Copyright 2021, Elsevier; (D) Unsupervised learning extended to Hofmann complexes, with ML-guided synthesis validated in Li||SPAN cells. Reproduced with permission from ref.[77]. Copyright 2025, Springer Nature; (E) Distribution of the lithium-ion-conductor dataset in the ICSD compositional space. Reproduced with permission from ref.[78]. Copyright 2023, Springer Nature; (F) Analysis of the electrochemical decomposition of LGPS. Reproduced with permission from ref.[79]. Copyright 2022, Wiley-VCH; (G) Workflow of high-throughput screening of Li-containing compounds in the MP and OQMD databases. Reproduced with permission from ref.[80]. Copyright 2025, Royal Society of Chemistry. DFT: Density functional theory; PCA: principle component analysis; XGB-C: XGBoost classifier; XGB-R: XGBoost regressor; ML: machine learning; ESW: electrochemical stability window; LGPS: Li10GeP2S12; SE: solid electrolyte; SVR: support-vector regression; LISICON: lithium super ionic conductor; MP: Materials Project; ICSD: Inorganic Crystal Structure Database; OQMD: Open Quantum Materials Database.
Algorithmic sophistication has rapidly increased with the development of target-driven pipelines and unsupervised learning. For garnet-type SEs, an AI-accelerated framework predicted migration barriers with a mean absolute error of 0.25 eV and achieved a computational speed nearly one billion times faster than ab initio methods. This approach screened 29,008 candidates and identified 12 predicted superionic conductors with RT conductivities up to 3.24 S cm-1 [Figure 4C][76]. Unsupervised clustering methods were subsequently introduced to map compositional landscapes without requiring labeled data. Applied to Li-ion conductors, these algorithms leveraged limited conductivity datasets to explore broad chemical spaces[81]. The same principle was extended to Hofmann-type complexes, where ML-guided synthesis optimized weakly coordinated Li+ environments, enabling 65% capacity retention after 500 cycles in Li|| sulfurized polyacrylonitrile (SPAN) cells [Figure 4D][77]. In parallel, attention-based models such as compositionally-restricted attention-based network (CrabNet) were trained on a curated dataset of 820 Li-ion conductors to classify high- and low-conductivity compounds, demonstrating how deep ML can extract latent compositional features from limited data [Figure 4E][78]. Reinforcement learning and deep Q-network-guided sampling further enabled efficient configuration searches for complex SEs such as Li6PS5Cl, achieving comparable accuracy to exhaustive enumeration but at a fraction of the computational cost[82]. Together, these approaches demonstrate that ML provides a scalable and data-efficient paradigm for high ionic conductor SE discovery.
Beyond ionic-conductivity prediction, ML has also been extended to evaluate the ESW, addressing limitations inherent to purely thermodynamic DFT approaches. Because decomposition involves both thermodynamic driving forces and kinetic accessibility, neither the direct-decomposition method (DDAM) nor the indirect-decomposition method (IDAM) alone is sufficient to capture realistic stability limits[83]. To resolve this, an integrated framework was proposed in which the electronic conductivities of all direct and indirect decomposition products are analyzed to determine the dominant decomposition pathway dynamically [Figure 4F][79]. This hybrid scheme more accurately distinguishes whether direct or indirect products govern electrochemical breakdown and shows good agreement with experimental measurements such as those for LGPS.
Building on this conceptual foundation, high-throughput ML workflows have been used to screen large databases for SEs with desirable ESW characteristics. A combined classification-regression approach identified candidates either predicted to be stable or exhibiting ESW ≥ 3 V. Applying this framework to the MP and Open Quantum Materials Database (OQMD) databases yielded 74 Li-containing SEs that satisfy thermodynamic stability, electronic insulation, practical feasibility, and ESW criteria [Figure 4G][80]. These results highlight that ML can substantially accelerate ESW assessment compared with DFT-only methods, although accuracy remains limited by the availability of high-quality reference DFT data and the scarcity of experimentally validated ESW measurements.
Feature- and physics-informed models
Composition-Structure Bimodal Network (COSNet) exemplifies the transition from handcrafted descriptors to learned multimodal representations by fusing composition and structure embeddings through Graph Convolutional Neural Networks (GCNs) and attention mechanisms, enabling robust property prediction even when structural data are limited [Figure 5A][84].
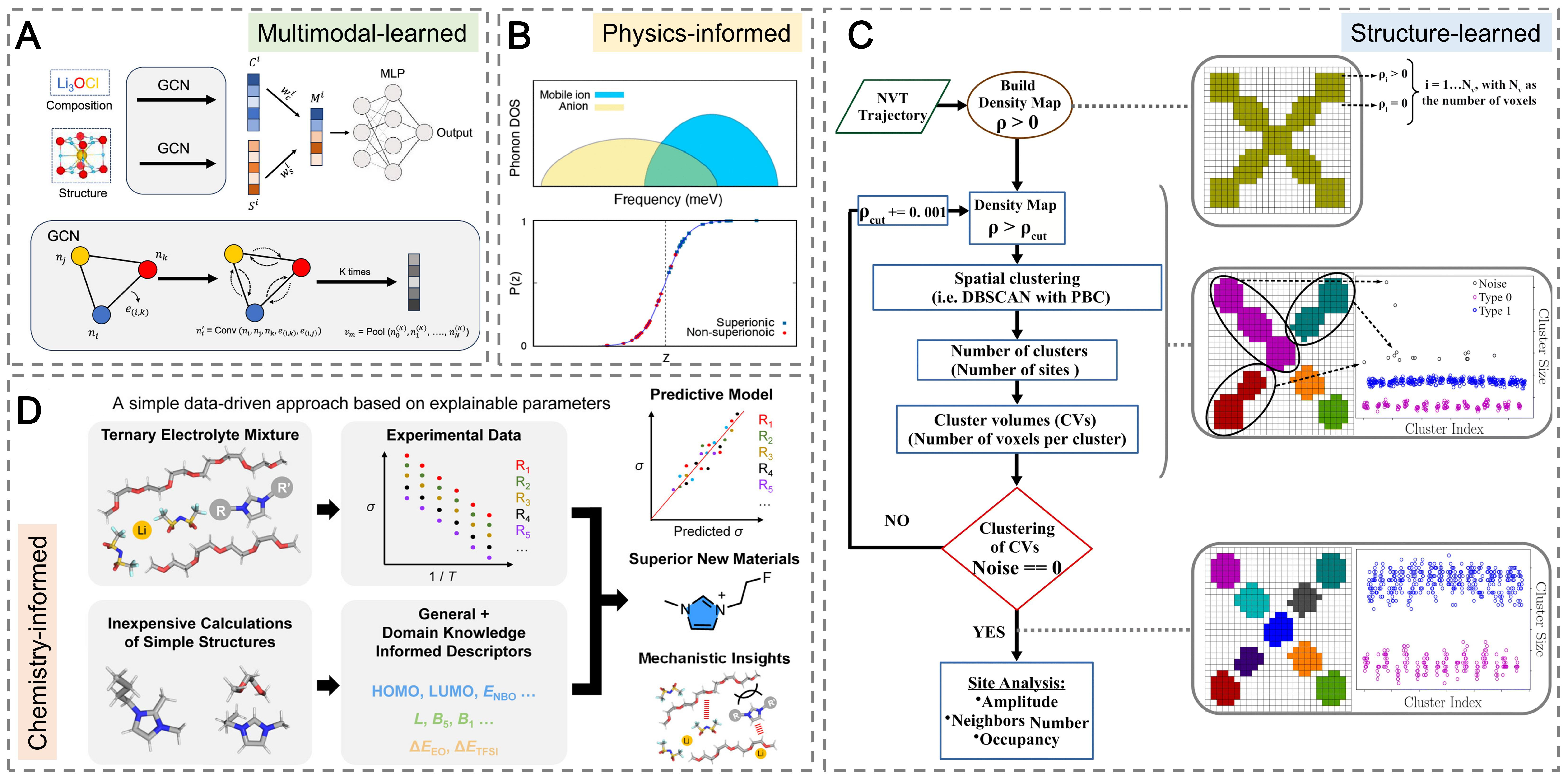
Figure 5. Representative descriptor paradigms enabling ML-driven SE discovery. (A) Multimodal-learned descriptors using the COSNet framework, where composition and structure graphs are encoded by GCNs and fused via attention into a unified representation for property prediction. Reproduced with permission from ref.[84]. Copyright 2024, American Chemical Society; (B) Physics-informed phonon descriptors for enhanced prediction of superionic conductors. Reproduced with permission from ref.[85]. Copyright 2024, American Chemical Society; (C) Structure-learned descriptors derived from graph neural networks (GNNs)-based clustering of Ga/Sc-doped LLZO, revealing dopant-site effects on Li+ transport. Reproduced with permission from ref.[86]. Copyright 2024, Royal Society of Chemistry; (D) Chemistry-informed descriptors capturing polymer-salt-IL interactions for random-forest prediction of ion conduction in solid polymer electrolytes. Reproduced with permission from ref.[87]. Copyright 2025, American Chemical Society. GCN: Graph convolutional neural network; PBC: periodic boundary conditions COSNet: Composition-Structure Bimodal Network; ML: machine learning; SE: solid electrolyte; LLZO: Li7La3Zr2O12; IL: ionic-liquid.
In parallel with these representation-learning advances, feature- and physics-informed models remain essential for interpretable design. In anti-perovskite SEs, ML identified a simple descriptor, t/η, that strongly correlates with ionic conductivity, leading to the discovery of nitro-halide double antiperovskites such as Li6NClBr2 and Li6NBrI2 with AIMD-validated ionic conductivities above 10-4 S cm-1[88]. Shi et al. developed a hierarchical encoding framework that uncovered 32 structural descriptors governing Li+ conduction in cubic argyrodites, thereby guiding the synthesis of Li5.5PS4.5Cl1.5 with an ionic conductivity of 9.4 × 10-3 S cm-1[89]. Simplified surrogate Hamiltonians, such as the pinball model, further reduced the computational cost of ion-dynamics simulations while retaining predictive accuracy[29]. Incorporating phonon-derived descriptors also improved the prediction of conductivity and diffusion coefficients by coupling lattice-vibrational and electronic features. The resulting model achieved 93% accuracy and was used to screen 264 Li-containing materials, identifying 11 promising candidates (e.g., Li8SnO6) as potential superionic conductors [Figure 5B][85].
Clustering analyses of Ga- and Sc-doped LLZO revealed that Li+ conductivity is maximized when Ga3+ preferentially occupies octahedral rather than tetrahedral sites, highlighting the role of dopant-site distributions in defining transport pathways [Figure 5C][86]. Extending from atomic-scale processes to fabrication behavior, Gaussian-process regression trained on shrinkage-temperature data up to 1,100 °C successfully extrapolated densification profiles to 1,200 °C with a root-mean-square error (RMSE) < 7 × 10-4, demonstrating that low-temperature data can reliably predict high-temperature sintering kinetics[90]. Polymer and interfacial systems represent emerging frontiers for ML-driven electrolyte design. In SPEs, coarse-grained MD combined with ML efficiently explored polymer–salt architectures, identifying formulations with RT ionic conductivities > 10-4 S cm-1[28]. Random forest regressors trained on curated datasets predict conductivity with a root-mean-square-error of 0.332 log(S cm-1)[91]. In particular, the inclusion of descriptors capturing non-covalent interactions between polymer chains, lithium salts, and ionic-liquid (IL) plasticizers significantly improved the model’s performance. The ML-model analysis revealed that ionic conduction in IL-plasticized SPEs is strongly governed by these intermolecular interactions, which determine ion mobility and segmental dynamics [Figure 5D][87]. Chemistry-informed neural networks embedding Arrhenius relations within message-passing architectures have predicted conductivities across thousands of SPE formulations[92].
ML methods have also advanced the understanding of interfacial and mechanical stability. Ahmad et al. combined graph convolutional networks, gradient boosting, and kernel ridge regression models trained on elastic constants to screen ~ 13,000 inorganic solids and more than 15,000 Li/SE interfaces for electrodeposition stability[27]. Their analysis revealed that mechanical anisotropy and lattice softness are key descriptors for dendrite suppression, providing quantitative design rules to balance interfacial stability and ionic transport. Lomeli et al. developed a model trained on structural and chemical descriptors from AIMD trajectories to classify 67 Li|SE interfaces as stable, passivating, or reactive[93]. The trained network rapidly predicted interfacial reactivity for thousands of compositions without additional simulations, identifying borate-based SEs such as LiB13C2 and LiB12PC as stable and self-passivating materials. These results illustrate how ML can bridge atomic-scale chemistry and macroscopic design rules.
ML potentials
Despite significant acceleration, descriptor-based regressors and surrogate Hamiltonians remain limited in accuracy and transferability, which motivates the development of MLIPs. MLIPs embed neural networks into potential-energy formulations, mapping atomic configurations to energies and forces with near-DFT precision, enabling simulations over much larger supercells and longer time scales[94,95].
Early MLIPs tailored to specific SE systems have demonstrated high fidelity and strong physical interpretability. For LLZO, a Deep Potential model trained on a combined database and DFT entries achieved total energies and atomic forces with high fidelity, capturing the tetragonal-to-cubic phase transition and realistic thermal expansion[Figure 6A][96]. In Li6PS5Cl, Behler-Parrinello-type neural-network potentials (NNPs) reproduced experimental conductivities (2-5 × 10-3 S cm-1 at RT) by incorporating disorder and avoiding high-temperature extrapolation, uncovering disorder-enhanced diffusion pathways and non-Arrhenius behavior that reconciled theory and experiment [Figure 6B][97]. These studies establish MLIPs as reliable tools for extracting mechanistic insight beyond the reach of conventional AIMD.
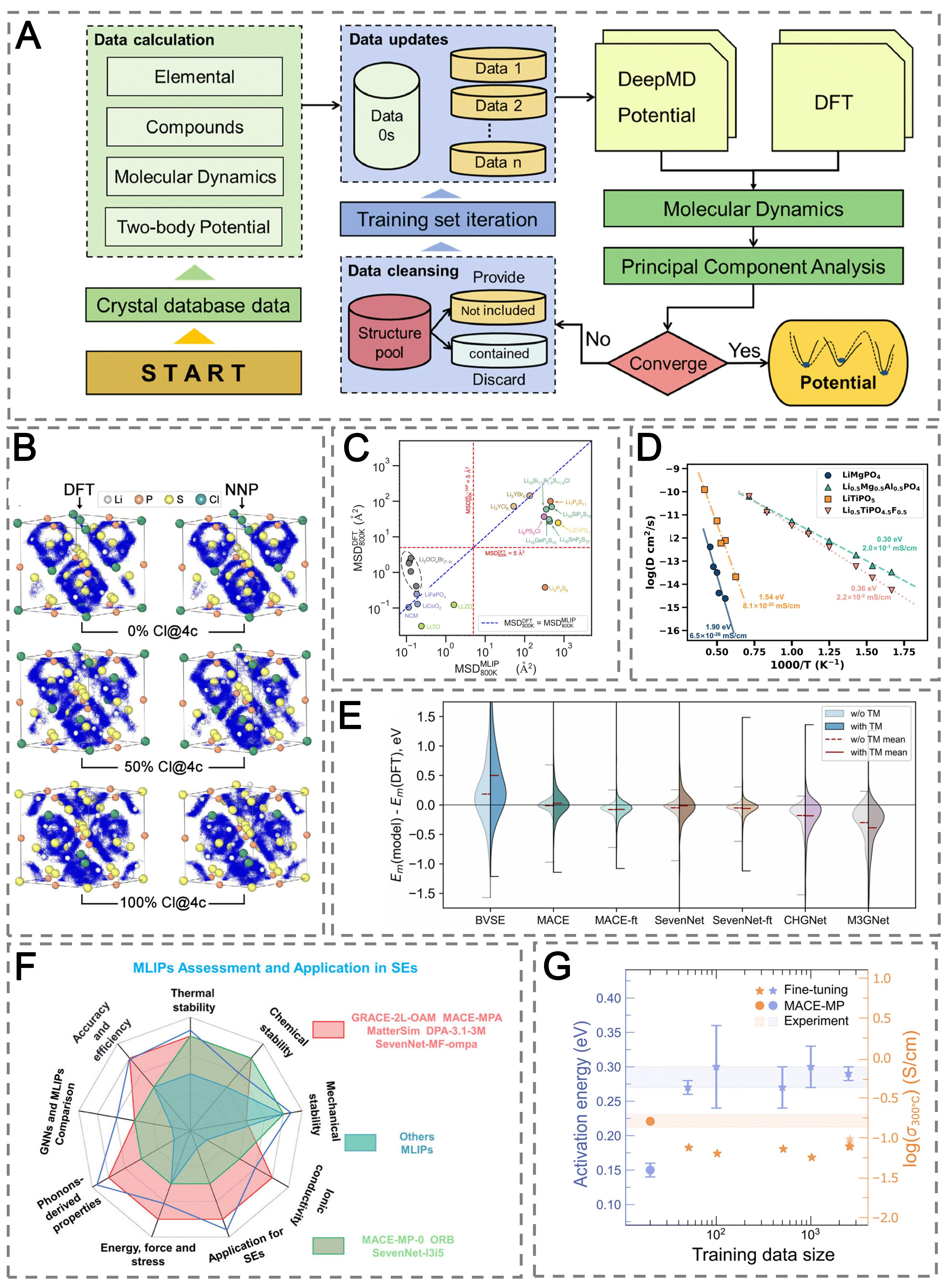
Figure 6. MLIPs for SE. (A) Training workflow and convergence evaluation of the LLZO deep-learning interatomic potential using principal component analysis for dataset coverage. Reproduced with permission from ref.[96]. Copyright 2024, Springer Nature; (B) Behler-Parrinello NNPs for Li6PS5Cl reproduce DFT accuracy and enable long MD simulations that capture disorder-enhanced Li+ diffusion. Reproduced with permission from ref.[97]. Copyright 2024, American Chemical Society; (C) MSDs comparison between MLIPs and DFT at 800 K for 18 well-known Li+ conductors. Reproduced with permission from ref.[98]. Copyright 2024, Royal Society of Chemistry; (D) Arrhenius plot of LiMgPO4, LiTiPO5, and their doped structures simulated by fine-tuned CHGNet-MD. Reproduced with permission from ref.[99]. Copyright 2025, Royal Society of Chemistry; (E) Violin plots of prediction errors for seven uMLIPs with and without transition metals benchmarked against DFT, Reproduced with permission from ref.[100]. Copyright 2025, Springer Nature; (F) Benchmarking and generalization of MLIPs across multiple models. Reproduced with permission from ref.[101]. Copyright 2025, American Chemical Society; (G) Fine-tuning and generalization of MACE-based MLIPs for activation energy and conductivity of Na3Zr2Si2PO12 at 300 °C. Reproduced with permission from ref.[102]. Copyright 2025, Royal Society of Chemistry. MD: Molecular dynamics; DFT: density functional theory; NNP: neural network potential; MSD: mean squared displacement; GNN: graph neural network; MACE: message passing atomic cluster expansion; SevenNet: scalable equivariance-enabled neural network; CHGNet: crystal hamiltonian graph neural network; M3GNet: materials GNN with 3-body interactions; GRACE: graph atomic cluster expansion; MP: Material Project; ORB: a family of uMLIPs for atomistic modelling of materials; SE: solid electrolyte; TM: transition metal; MLIP: machine-learning interatomic potential; SE: solid electrolyte; LLZO: Li7La3Zr2O12; uMLIP: universal MLIP.
Building on system-specific successes, MLIPs have also been integrated into high-throughput workflows. An ML-accelerated funnel screened ~ 7 × 105 Li-containing crystals using a universal graph-based potential as a DFT surrogate, discarded mechanically or electronically unsuitable phases, and then applied subsequent MD filtering to identify fast-ion conductors. Roughly 130 candidates remained, including ~ 28 halides such as KLi2ScBr6 and Rb2Li3Br5, projected to approach an ionic conductivity of 1 × 10-2 S cm-1 at RT while maintaining shear modulus above 8 GPa [Figure 6C][98].
Beyond system-specific models such as neural equivariant interatomic potentials (NequIP)[103], Deep Potential[104], and TensorMol (a neural-network interatomic potential incorporating long-range electrostatics and van der Waals interactions)[105], recent efforts have focused on developing uMLIPs trained on large, chemically diverse datasets (e.g., MP)[106]. Representative models include CHGNet[107], M3GNet[108,109], and -MP-0 (a uMLIP trained using the MACE framework on the MP database)[110], which extend applicability across multiple chemistries. A CHGNet-based nudged elastic band (NEB) framework fine-tuned with DFT-calculated transition states achieved near-DFT fidelity in mapping migration barriers for NASICON-type electrolytes and identified new Pnma-type conductors such as LiMgPO4 and LiTiPO5 [Figure 6D][99]. Building on these structural prototypes, doped variants such as Li0.5Mg0.5Al0.5PO4 and Li0.5TPO4.5F0.5 were further examined and were found to exhibit ionic conductivities of 0.20 mS cm-1 and 0.022 mS cm-1, respectively.
Despite their broad applicability, uMLIPs exhibit notable limitations when extrapolating to chemistries insufficiently represented in their training distributions. Benchmark studies consistently show that uMLIPs systematically underestimate Li+ migration barriers by ~ 150 meV relative to DFT, with larger errors observed in transition-metal-containing systems and highly disordered structures. To systematically quantify these errors, benchmark datasets such as LiTraj (Datasets for benchmarking ML models for predicting Li-ion migration) provide standardized validation resources across diverse Li-host materials. Using LiTraj, classical ML models and GNNs accurately distinguish fast from poor ionic conductors and predict migration barriers with near-DFT accuracy [Figure 6E][100]. Comparative benchmarks of twelve MLIPs, including graph atomic cluster expansion (GRACE), deep potential with attention (DPA), a deep learning atomistic model across elements, temperatures and pressures (MatterSim), MACE, scalable equivariance-enabled neural network (SevenNet), CHGNet, a neural network framework based on Cartesian tensor representations for molecular potential learning (TensorNet), M3GNet, and a family of uMLIPs for atomistic modelling of materials (ORB), covering energy/force accuracy, thermodynamics, elasticity, and Li+ diffusivity [Figure 6F][101]. Models such as a two-layer GRACE uMLIP model trained on Open Material 2024 (OMAt24) dataset and fine-tuned on subsampled Alexandria (sAlex) and Materials Project Trajectory (MPtraj) datasets (GRACE-2L-OAM), a uMLIP trained using the MACE framework on the MPTraj and sAlex datasets (MACE-MPA), MatterSim, and a SevenNet-based GNN interatomic potential trained using multi-fidelity learning on the combined OMat24, MPTraj, and sAlex datasets (SevenNet-MF-ompa) achieved robust performance. MatterSim-based simulations indicated that 40%-50% S/Cl anion disorder in Li6PS5Cl enhances migration connectivity, while Li-rich compositions in Li3YCl6 expand conduction channels and reduce diffusion barriers. These errors have been attributed to domain-shift effects and a softening of the learned potential-energy surface, underscoring the intrinsic challenges of universal generalization.
Addressing these limitations requires adaptive strategies that combine targeted data acquisition with model refinement. Active-learning schemes that prioritize uncertain or high-energy configurations, together with few-shot or task-specific fine-tuning, have proven particularly effective. For example, retraining foundation models such as MACE-MP-0 on fewer than ~ 300 targeted configurations restores migration-barrier accuracy in LiF to within ~ 0.02 eV of DFT[111], while iterative fine-tuning of CHGNet captured composition-dependent conductivity trends in Li3YCl6-xBrx halide SEs, achieving quantitative agreement with experimental diffusion coefficients[112]. The success of fine-tuning workflows across halide, sulfide, and oxide families demonstrates the adaptive specialization of uMLIPs without retraining from scratch. MACE-based MLIPs have been further generalized to cover SEs such as Na1+xZr2SixP3-xO12 and Li3+xP1-xGexS4-4xO4x using representative training sets [Figure 6G]. Applications to mixed halides identified Li3In0.5Y0.5Br3Cl3 as a composition with favorable migration pathways[102]. Continual-learning frameworks such as replay and elastic weight consolidation (reEWC) combine experience replay and elastic weight consolidation to improve Li-ion diffusivity and potential-energy accuracy while preventing catastrophic forgetting[113]. Beyond task-specific retraining, systematic analyses have shown that pretrained uMLIPs often exhibit a potential-energy-surface softening effect, leading to underestimated migration barriers and defect energetics; this bias can be effectively corrected through fine-tuning on a limited number of high-energy configurations[114]. At the infrastructure level, standardized fine-tuning workflows based on foundation models are increasingly adopted across materials classes, including SEs, defects, and heterointerfaces, improving model reliability and transferability[115]. In addition to improving accuracy, MLIPs have also enabled new physical insights; for example, by identifying machine-learned structural fingerprints (“softness”) that correlate local disorder motifs with enhanced room-temperature ionic conductivity in thiophosphate electrolytes[116].
Collectively, these results demonstrate that uncertainty-aware retraining, active learning, and few-shot adaptation are essential for extending uMLIP reliability across unseen chemistries, positioning MLIPs as a cornerstone of scalable and adaptive SE discovery workflows
AI AGENTS FOR SE DISCOVERY
The emergence of AI agents around 2025 has begun to mark a methodological turning point in SE research. Unlike conventional ML models operating static workflows, AI agents feature autonomy, adaptability, and closed-loop integration, enabling perception of heterogeneous data, reasoning and hypothesis generation, experimental action, and learning from feedback. These capabilities transform AI from a passive predictor into an active collaborator in SE discovery. Representative AI agent platforms differ in autonomy, experimental coupling, and targeted SE tasks, and collectively span different stages of the agent loop, from knowledge perception and reasoning to autonomous experimental action, as summarized in Table 1.
Representative AI agent platforms for SE discovery across perception, reasoning, and autonomous experimentation
| Platform | Category | Autonomy level | Experiment coupling | Database quality | Primary SE-related role | Key limitation | Ref. |
| DigBat (DDSE) | Data-driven SE framework | High | No | High (experimentally curated with manual validation) | Data-driven SE screening and mechanistic insight | Without an autonomous synthesis system | [117] |
| ChatSSB | Domain-specific RAG agent | Low-Medium | No | Medium-High (domain-curated SSB literature corpus) | SSB-focused assistant for literature mining and planning | Limited autonomy; depends on corpus and ontology quality | [118] |
| Uni-Electrolyte agent | Knowledge-integration agent | Medium | Indirect | Medium-High (integrated literature and DFT databases) | Electrolyte/SEI co-design via literature + DFT databases + ML predictors | Metadata fragmentation; schema incompatibility | [119] |
| DIVE | Multi-agent perception and reasoning system | Medium-High | No | High (large-scale, literature-curated from figures and tables) | Data extraction and inverse design for hydride-based SE | Currently limited to hydrogen-containing materials | [120] |
| CRYSTAL | Multi-agent reasoning system | Medium | No | Curated external databases | General phase-diagram/phase-mapping reasoning; supports compositional navigation for SE candidates | Weak linkage to processing constraints | [121] |
| ChemCrow | Tool-using reasoning and planning agent | Medium | No | Not applicable (tool-using LLM* without native database) | General chemistry planning/protocol agent | Requires human validation; tool/constraint dependence | [122] |
| ORGANA | Robotic execution agent | Medium | Yes (human-supervised) | Not applicable (execution-focused platform) | General robotic execution of multistep chemistry | Inert-atmosphere integration remains challenging | [123] |
| A-Lab | Autonomous action and learning agent | High | Yes | Indirect (literature-mined synthesis rules + DFT databases) | Closed-loop synthesis and validation of inorganic solids | Limited atmosphere control; mainly powder synthesis | [124] |
Literature and text mining
As summarized in Table 1, literature and text-mining agents are among the most mature agent paradigms, providing structured knowledge that supports downstream discovery and autonomous experimentation. Early efforts in literature mining relied primarily on NLP and ML to extract structured information from the rapidly growing corpus of materials science literature. These systems compiled synthesis parameters, processing conditions, and electrochemical properties for sulfide- and oxide-based SEs, revealing, for example, low-temperature LLZO synthesis routes that mitigate interfacial degradation during cell assembly [Figure 7A][125]. Large-scale NLP pipelines further extracted ionic conductivity values from over 3,200 publications, producing databases with far higher coverage and consistency than manual curation. This automated pipeline distinguished ionic from electronic conductivity, identifying 68.7% of previously ambiguous records as ionic and thereby reducing the proportion of unknown entries from 93% to 24.3% [Figure 7B][126].
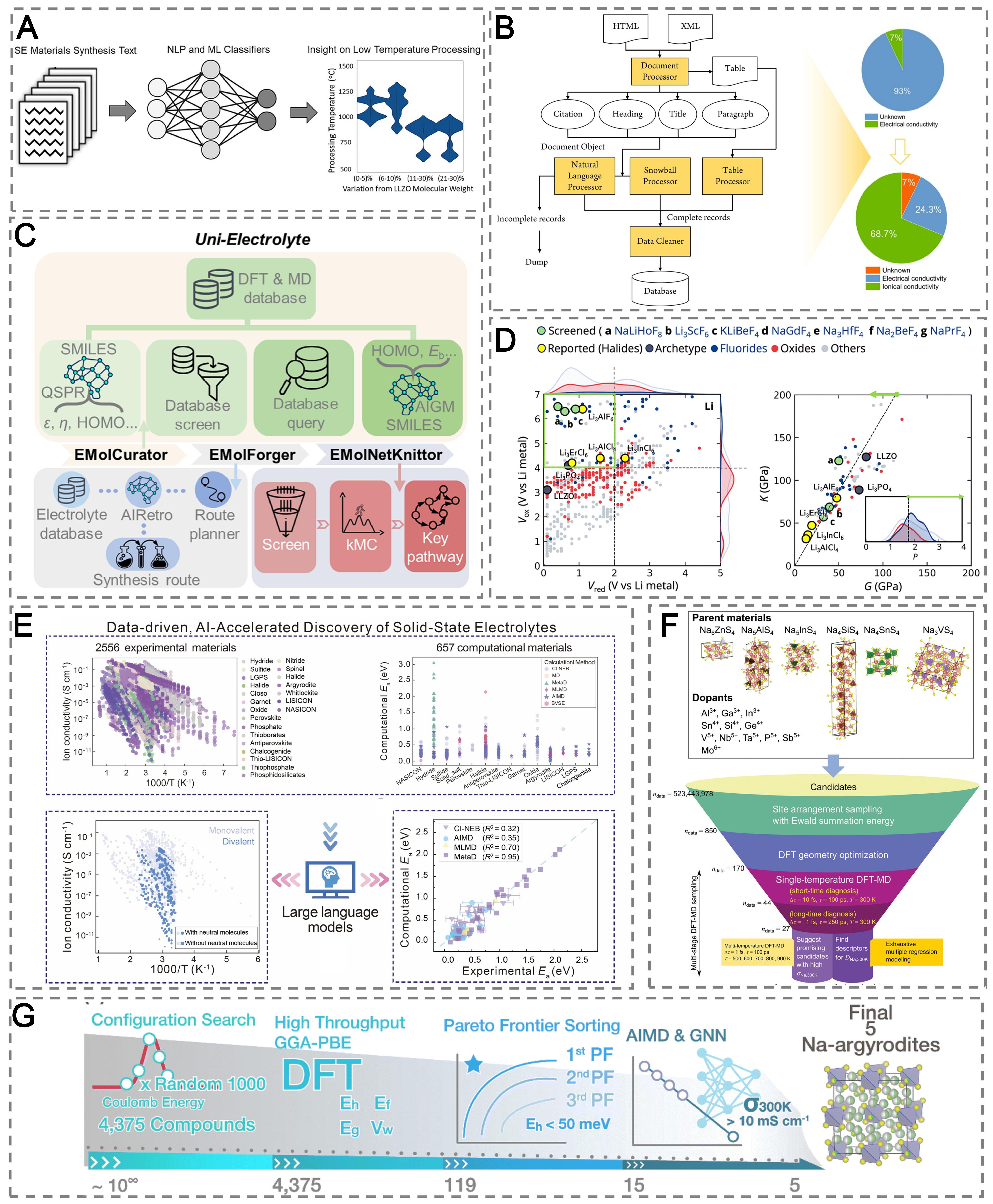
Figure 7. AI agents for knowledge mining and data-driven discovery of SE. (A) Text-mining framework identifying low-temperature processing routes for garnet-type electrolytes. Reproduced with permission from ref.[125]. Copyright 2020, Elsevier; (B) Automated NLP pipeline extracting ionic conductivity values from > 3,000 literature and the application. Reproduced with permission from ref.[126]. Copyright 2023, American Chemical Society; (C) Uni-Electrolyte agent integrating literature mining, computational databases, and kinetic modeling for closed-loop electrolyte design. Reproduced with permission from ref.[119]. Copyright 2025, Wiley-VCH; (D) Data-mining workflow for fluoride-based SEs combining electrochemical and mechanical screening. Reproduced with permission from ref.[127]. Copyright 2024, Royal Society of Chemistry; (E) Integrated LLMs to reveal mechanisms in SE based on the DDSE database. Reproduced with permission from ref.[117]. Copyright 2025, Wiley-VCH; (F) High-throughput discovery of Na-based sulfides using crystallographic databases and multi-stage DFT-MD simulations, uncovering three solid-solution series with high Na+ conductivity. Reproduced with permission from ref.[128]. Copyright 2022, Wiley-VCH; (G) A schematic representation of the computational screening sequence for Na-argyrodite SEs. Reproduced with permission from ref.[129]. Copyright 2025, Royal Society of Chemistry. SE: Solid electrolyte; NLP: natural language processing; ML: machine learning; DFT: density functional theory; MD: molecular dynamics; AIMD: ab initio molecular dynamics; GNN: graph neural network; AI: artificial intelligence; LLM: large language model; DDSE: Dynamic Database of Solid-state Electrolytes.
While such pipelines accelerated data collection, they operated as static workflows lacking reasoning or adaptability. Recent progress has transformed these systems into dynamic AI agents capable of perceiving multimodal information, reasoning across sources, and updating knowledge through feedback. For instance, Chen et al. summarized the AI agent capable of extracting conductivity descriptors, electrochemical stability limits, and interfacial reaction patterns to construct evolving knowledge graphs for electrolyte design[130]. Multimodal agents combine vision and language models to parse figures and tables directly, reducing manual data-extraction time by more than 80%[131,132].
A recent representative example is the Descriptive Interpretation of Visual Expression (DIVE) framework, which moves beyond single-model multimodal extraction toward a coordinated multi-agent perception and reasoning pipeline. Instead of directly extracting values from figures, DIVE decomposes graphical elements into semantic primitives, cross-validates extracted information across figures, tables, and text, and incrementally constructs a structured database with provenance tracking. Applied to hydrogen-containing solid materials, this workflow curated over 30,000 data entries from more than 4,000 publications and enabled rapid inverse design of previously unreported hydride compositions[120].
Beyond figure parsing, recent multimodal AI agents increasingly integrate structural and spectroscopic data with textual knowledge to enable hypothesis generation rather than passive information retrieval. For example, autonomous characterization agents have been developed to interpret X-ray diffraction (XRD) data in self-driving laboratory settings, providing probabilistic phase identification and uncertainty-aware structural assignments that directly support inverse design and robotic discovery workflows[133]. Beyond text mining itself, higher-level reasoning agents can operate on knowledge extracted from literature and databases. At a higher level of reasoning, multi-agent systems such as a multi-agent AI system for crystal-structure phase mapping (CRYSTAL) integrate crystallographic databases, thermodynamic constraints, and learning-based inference to automatically generate physically meaningful phase diagrams, thereby enabling the discovery of previously inaccessible materials and guiding hypothesis-driven exploration of compositional spaces[121].
Expanding beyond information extraction, integrated systems such as the Uni-Electrolyte agent couple literature mining with DFT-based databases and ML property predictors to support the rational design of electrolyte compositions and solid-electrolyte interphase (SEI) chemistries [Figure 7C][119]. However, without findable, accessible, interoperable, and reusable (FAIR)-aligned data practices, these agents still face fragmented schemas and incompatible metadata. In particular, ontology gaps in synthesis parameters and disorder metrics limit cross-lab transfer and agent-level reasoning.
Data-driven discovery and optimization
Building on knowledge-mining agents [Table 1], data-driven discovery and optimization agents integrate databases, physics-based models, and learning algorithms to navigate the compositional and processing design space of SEs. Before the advent of ML, heuristic and physics-based models such as the bond valence sum (BVS) approach provided an efficient means to estimate ion migration barriers and screen potential fast-ion conductors. Wong et al.[134] and Chen et al.[135] developed the soft bond valence (softBV) framework, where activation energies are derived from local bond valence environments and electrostatic interactions between mobile ions and their counterions[134,135]. Despite its simplicity, the BVS model shows good agreement with DFT-calculated barriers and has been incorporated into high-throughput screening workflows to identify halide-type ion conductors[136,137]. The physical interpretability and low computational cost of such models make them valuable references for developing AI-based descriptors or validating “black-box” predictions.
Building on these physically grounded foundations, data-driven approaches have accelerated the discovery of SEs by extending design objectives beyond ionic transport to include mechanical deformability and interfacial stability. Systematic screening of the MP database, combined with ML-accelerated MD, identified ternary fluorides such as Li3ScF6, NaPrF4, and Na3HfF7 as promising SEs exhibiting favorable diffusivity, interfacial stability, and mechanical softness [Figure 7D][127]. Complementary elasticity-based models trained on more than 14,000 structures achieved high predictive accuracy for shear and bulk moduli. An active-learning strategy further improved coefficient of determination (R2) scores by 32%-63% with limited new data, demonstrating how adaptive sampling accelerates multi-objective optimization[138].
AI agents refer to integrated frameworks that combine data resources, learning models, and reasoning modules to guide discovery, rather than standalone predictive models. In this sense, agent-enabled discovery increasingly depends on large, heterogeneous, and continuously evolving datasets. A representative effort is the Dynamic Database of Solid-state Electrolytes (DDSE), which has now been further developed into the DigBat platform (www.digbat.org). By 2025, DDSE had compiled ionic-conductivity data for over 3,000 SEs (exceeding 20,000 measurements) across. Li+, Na+, K+, Ag+, Ca2+, Mg2+, and Zn2+ systems[139,140], representing one of the largest curated databases of SEs to date. Analyses of DDSE revealed the under-explored potential of divalent-ion conductors, providing quantitative guidelines for next-generation multivalent electrolytes. Building on DDSE, one of the earliest AI-agent frameworks introduced in solid-state-battery research was developed by coupling this database with large language models (LLMs) and ab initio metadynamics simulations. This framework uncovered a two-step ion-migration mechanism in hydride conductors, in which molecules mediate unconventional hopping pathways [Figure 7E][117]. More importantly, this framework revealed fundamental limitations of conventional ion-migration simulations (i.e., CI-NEB and AIMD) and demonstrated that ab initio metadynamics provides a more reliable description of cation transport.
Large-scale exploration of Na-based sulfide SEs has further leveraged crystallographic repositories such as ICSD[141], MP[106], and Springer Materials. Focusing on tetrahedral frameworks analogous to Na3SbS4, multi-stage DFT-MD sampling generated 105 trajectories and revealed three solid-solution series with RT Na+ conductivities up to 10-2 S cm-1 [Figure 7F][128]. In Na-based argyrodites, Merchant et al. expanded this approach by integrating Google DeepMind[142] and established an integrated DFT-AIMD-ML workflow. A compositional space of 4,375 hypothetical Na argyrodite structures was systematically explored, and key thermodynamic and electrochemical descriptors, including energy above the hull (Eh), formation energy (Ef), band gap (Eg), and ESW (Vw), were predicted using Connectivity-Optimized Graph and Nested Graph Networks (coGN and coNGN). Subsequent AIMD simulations identified five promising Na argyrodites, including Na6SiS4Cl2, which exhibited a theoretical RT ionic conductivity of 2.9 × 10-2 S cm-1 [Figure 7G][129].
Moving toward autonomous discovery, Chen et al. combined a cloud-scale AI workflow for computational screening with autonomous synthesis to identify new halide SEs[143]. This workflow explored over 107 compositions, prioritized Li/Na mixed-halide candidates, and experimentally validated NaxLi3-xYCl6 (e.g., Na2LiYCl6) with enhanced ionic conductivity and stability, thereby completing the model-to-experiment cycle. Statistical roadmaps derived from these datasets further highlighted halides and nitrides as promising families. They also emphasized the importance of kinetic stabilization, alongside thermodynamic stability, in achieving Li-metal compatibility[144].
In a complementary direction, the integration of GPT-4o with representation clustering has opened a new avenue for discovering metal-organic framework (MOF)-based SEs. By mining over 11,000 MOF candidates, extracting structural-electrochemical descriptors, the workflow identified a unique candidate MOF material (NOTT-400) as a previously unreported Li+ conductor (2.23 × 10-4 S cm-1) with a wide ESW (0-4.79 V), validating the predictive workflow experimentally[145]. This approach exemplifies LLM-assisted materials discovery, bridging unstructured knowledge extraction with data-driven design and marking a step toward generalized AI agents capable of reasoning across materials domains.
Beyond database-driven screening, generative AI is emerging as a new paradigm for exploring uncharted compositional and structural spaces in SE. Nguyen et al. introduced a physics-informed hierarchical generative framework, termed SHAFT-density (symmetry-aware hierarchical architecture for flow-based traversal with density), which integrates crystallographic symmetry principles with reinforcement learning[146]. By embedding empirical physical constraints such as lattice symmetry, packing density, and formation energy, the framework efficiently generates chemically valid and structurally stable candidates. It successfully identified metastable halide phases (e.g., LiBr, LiCl, Li2IBr, and Li3CBr2) with favorable ionic conductivities and thermodynamic stabilities, expanding the design window beyond the existing database. SHAFT exemplifies how physics-informed generative models complement data-mining workflows, enabling autonomous and physically interpretable exploration of SE chemistries.
Autonomous experimentation
As outlined in Table 1, autonomous experimentation agents represent the highest degree of integration, coupling planning and decision-making with robotic execution and online characterization to enable closed-loop SE optimization. SE represents a complex and highly constrained search space, where composition, processing, and interfacial factors interact in nonlinear ways. Evaluating these interactions is both costly and difficult to generalize from limited data. Autonomous experimentation addresses this challenge by integrating robotic execution, online characterization, and AI-driven decision-making to explore the design spaces efficiently under controlled atmospheres and standardized protocols[147].
Self-driving laboratory platforms illustrate the principle of closed-loop experimentation. Integrated systems combine automated synthesis and in-line diffraction with control software that updates plans as new evidence accumulates. These systems minimize the delay between synthesis and analysis and ensure reproducible workflows with full traceability of decisions. Language and vision-based agents further connect planning tools and instruments by retrieving prior knowledge, generating experimental templates, and automatically producing safe operating procedures[148]. Extending this concept to general laboratory automation, ORGANA (an assistive robotic system designed to support fundamental chemistry experiments, bridging important advancements in robotics and planning with the chemistry community) serves as an assistive robotic system that automates complex chemical experiments through AI-based perception and decision-making. Integrating vision and language models with task and motion planning allows robots to execute multistep workflows while maintaining real-time communication with researchers[123]. An autonomous laboratory (A-Lab) extends these capabilities to solid-state systems, demonstrating the integration of simulation, ML, and robotics toward autonomous materials discovery. In 17 days of continuous closed-loop operation, the platform performed 355 experiments and synthesized 41 previously unreported inorganic compounds, achieving a 71% success rate. This performance was achieved by coupling DFT-guided target selection with text-mined synthesis rules, ML interpretation of experimental data, and active-learning refinement of failed reactions. The results demonstrate that autonomous research agents can rapidly translate computational predictions into experimental realization, offering a scalable framework for accelerating SE development [Figure 8A][124].
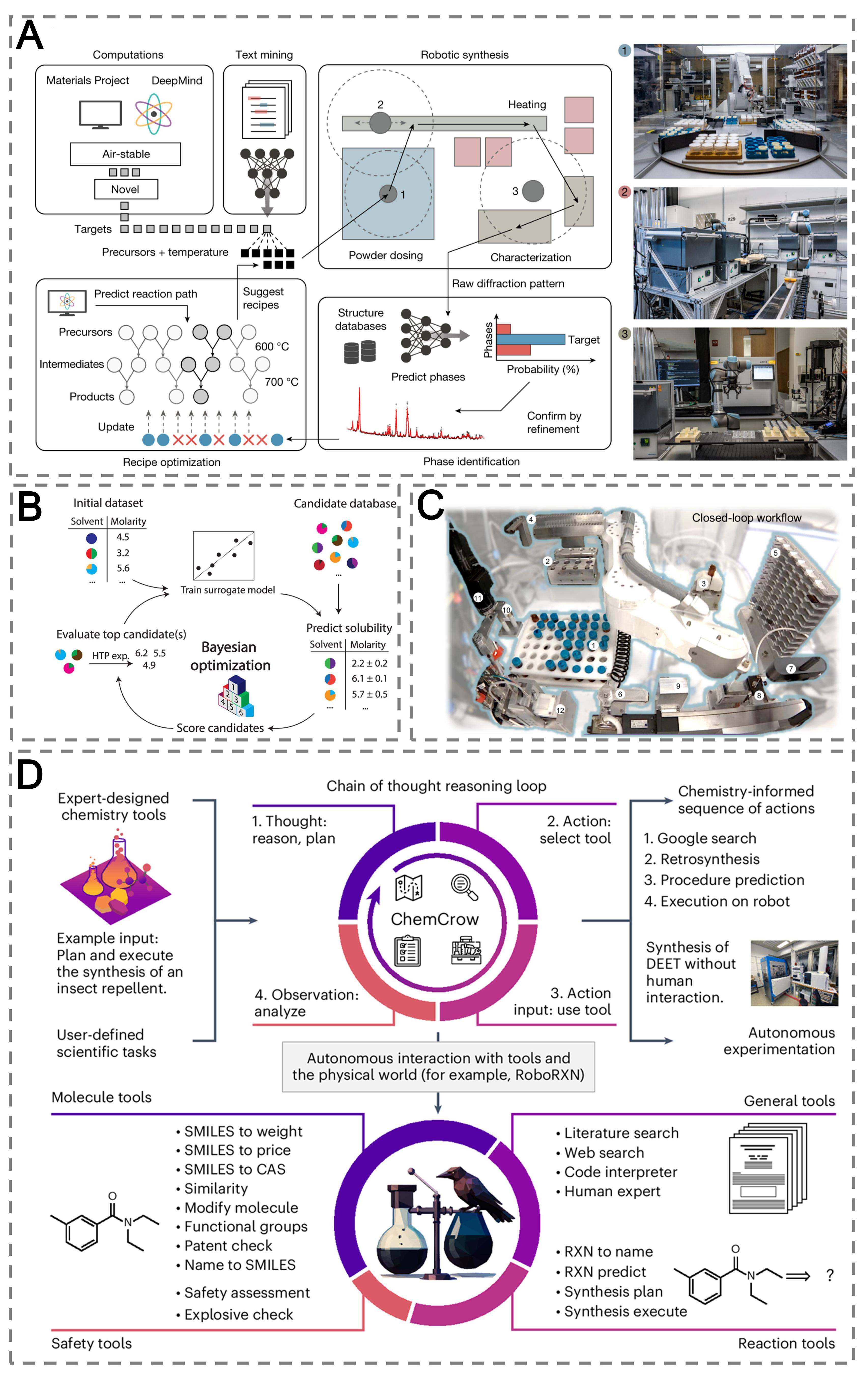
Figure 8. AI agents and data-driven frameworks for autonomous experimentation in solid material research. (A) A-Lab, a self-driving laboratory integrating computation, text mining, robotic synthesis, and automated characterization into a closed-loop workflow for solid materials discovery. Reproduced with permission from ref.[124]. Copyright 2023, Springer Nature; (B) Schematic of the BO algorithm for accelerated screening of binary solvents with targeted anolyte solubility. Reproduced with permission from ref.[149]. Copyright 2024, Springer Nature; (C) (1) solution storage rack, (2) solution heating and mixing module, (3) capping and uncapping system, (4) pipette rack, (5) substrate rack, (6) substrate gripper, (7) imaging station, (8) blade-coating station, (9) blade cleaning station, (10) annealing station, (11) thickness characterization station, (12) electrical characterization station. Polybot platform for automated fabrication and optimization of electronic thin films. Reproduced with permission from ref.[150]. Copyright 2025, Springer Nature; (D) ChemCrow, an LLM-based chemistry agent that leverages multiple expert-designed tools to autonomously plan and execute chemical syntheses. Reproduced with permission from ref.[122]. Copyright 2024, Springer Nature. HTP: High-throughput; CAS: Chemical Abstracts Service; DEET: N,N-Diethyl-meta-toluamide, (Robo)RXN: IBM RXN for Chemistry; SMILES: simplified molecular input line entry system; A-Lab: an autonomous laboratory; AI: artificial intelligence; BO: Bayesian optimization; LLM: large language model.
Active learning provides a general framework for adaptive experiment selection, in which Bayesian optimization (BO) serves as the dominant strategy when evaluation costs are high. A probabilistic surrogate model, often based on Gaussian processes, is trained on collected data to predict objectives and associated uncertainties[151]. An acquisition function identifies the most informative next candidates by balancing exploration and exploitation. After each experimental cycle, the model is updated and proposes new experiments [Figure 8B][149,152]. This adaptive process fits composition and processing studies and can be extended to multi-objective optimization to define relationships among ionic conductivity, interfacial impedance, and processability.
Applications in both solid and liquid electrolytes demonstrate the efficiency of this approach. In aliovalent-doped NASICONs, BO reduced the search space by almost 80% and identified compositions such as Li1.3Ca0.1Zr1.9Si0.1P2.9O12 with ionic conductivities above 3.1 × 10-5 S cm-1[153]. Statistical analysis indicated that dopant concentration and heat treatment were dominant factors influencing conductivity[154,155]. Similar workflows have been applied to liquid electrolytes. In rechargeable aprotic Li-O2 batteries, automated high-throughput robotic experiments coupled with BO rapidly identified electrolyte formulations that improved dis/charge efficiencies, achieving stable cycling over 100 cycles at 0.5 mAh cm-2[156]. Robotic platforms have also screened hundreds of aqueous and non-aqueous electrolytes within hours, achieving high precision (ionic conductivity ≈ 0.5 × 10-3 S cm-1, ΔV ≈ 0.02 V) and reproducibility[157]. Dave et al. integrated BO with a robotic test stand to screen 140 aqueous electrolyte formulations within 40 hours, discovering a non-intuitive mixed-anion Na electrolyte with enhanced stability and releasing a curated dataset of 251 compositions[158].
Integration of robotics, ML, and online measurement is further expanding autonomous electrolyte optimization. The Clio-Dragonfly platform couples a robotic unit with a BO planner to refine lithium-ion electrolyte formulations, completing 42 experiments in 2 days and discovering 6 formulations with improved fast-charging performance[62]. Automated systems for polymer electrolytes can prepare and test more than 60 samples per researcher hour, generating the largest unified dataset for Li+ and Na+ conduction in PEO-based electrolytes[159]. Such datasets accelerate SPEs optimization and improve the training of ML models for hybrid or composite SEs.
Closed-loop optimization has also been extended to polymer processing platforms equipped with automated modules for solution handling, coating, annealing, imaging, and electrical characterization [Figure 8C][150]. These systems enable rapid exploration of multidimensional parameter spaces under multi-objective optimization. Although originally designed for conductive polymer films, the same workflow can be adapted for polymer-salt mixtures. For SEs, dry-room operation, compatible substrates, and impedance-based workflows are required to measure conductivity and activation energy while ensuring film uniformity and minimizing defect density. Defining feasible processing limits, including viscosity range, film integrity, and thermal budget, can further enhance reproducibility and accelerate convergence.
Despite these advances, several practical bottlenecks currently limit the deployment of autonomous laboratories for SE research, particularly for air-sensitive halide electrolytes. Halide SEs such as Li3YCl6 and Li3InCl6 exhibit high ionic conductivity and favorable electrochemical stability, yet their pronounced sensitivity to moisture and oxygen necessitates strict inert-atmosphere handling, typically relying on glovebox or dry-room environments[14,57]. Integrating such controlled environments with robotic platforms remains technically challenging and costly, as sealed material transfer, continuous atmosphere monitoring, and contamination control must be maintained throughout the entire synthesis and characterization workflows.
In addition, most existing autonomous platforms focus on powder synthesis and screening, whereas industrially relevant halide electrolytes often require scalable processing into dense pellets or thin films with controlled microstructure and interface quality. Automating multistep fabrication processes such as pressing, sintering, coating, and cell assembly for mechanically soft halide materials remains nontrivial and has been only sparsely demonstrated[56]. Moreover, high capital and maintenance costs associated with robotic hardware, glovebox-compatible instrumentation, and in situ characterization further limit widespread adoption of such systems.
Potential mitigation strategies include modular robotic designs compatible with dry-room operation, hybrid human-robot workflows for particularly sensitive handling steps, and the use of data-efficient, agent-guided experiment planning to minimize the total number of required experiments. Incorporating materials-specific constraints, such as moisture tolerance windows and processing limits, into autonomous decision-making frameworks may further improve robustness and accelerate translation from laboratory-scale discovery to manufacturable halide SEs.
Advancing autonomous experimentation for SE requires progress along three key directions. First, the integration of specialized in situ probes such as XRD for phase identification, temperature-controlled impedance cells, and automated assembly tools for electrode and electrolyte interfaces will ensure that each experimental iteration yields mechanistic insights rather than single-point outcomes. Second, the implementation of multi-objective BO with well-defined constraints can simultaneously optimize ionic conductivity, electrochemical stability, interfacial impedance, and pellet density under realistic synthesis conditions. Third, the development of agent-based control systems that couple literature-derived priors, thermochemical rules, and safety protocols with electronic laboratory notebooks and data repositories is essential to enhance reproducibility and data reuse [Figure 8D][122].
Recent progress highlights the evolution from domain-specific assistants to general-purpose scientific agents capable of reasoning, planning, and execution. ChemCrow integrates LLMs with expert-designed tools to autonomously plan and perform chemical syntheses, bridging text-based reasoning and laboratory execution. Building on this concept, Coscientist, a GPT-4-powered system, achieves a closed reasoning-planning-execution loop by combining language models with web retrieval, code execution, and robotic automation. It successfully optimized palladium-catalyzed cross-coupling reactions, demonstrating how foundation-model-driven agents can translate high-level objectives into executable workflows and iteratively refine them through feedback[160].
Meanwhile, domain-specific intelligent agents are also emerging to accelerate materials research. An LLM-based intelligent research assistant specifically designed for solid-state battery research (ChatSSB), a retrieval-augmented generation (RAG) system tailored for solid-state batteries (SSBs), integrates multi-agent collaboration, expert feedback, and analytical tools to enable precise literature mining, data visualization, and experimental planning. Applied to LLZO electrolytes, ChatSSB demonstrates how specialized AI assistants can bridge knowledge extraction and autonomous experimentation, enhancing efficiency and reproducibility in battery materials discovery[118].
Autonomous laboratories thus represent a continuum from automated experimentation to cognitively integrated AI agents. When coupled with uncertainty-aware selection, standardized measurement schemes, and interoperable data infrastructures, they offer an auditable foundation for accelerating SE discovery.
Multiscale modeling and integration
Multiscale modeling provides a unified framework linking atomic-scale processes with mesoscale and continuum behavior, thereby offering mechanistic insights into ion transport, stress evolution, and failure in SEs. Recent studies have integrated quantum-level calculations, atomistic simulations, and continuum methods to describe the coupled electrochemical and mechanical responses that govern degradation during repeated cycling. Among these approaches, phase-field modeling has become a powerful tool for simulating dendrite formation and chemo-mechanical stress evolution in SEs. By representing diffuse interfaces and evolving microstructures, phase-field models capture morphological instabilities that arise during electrochemical operation and describe the interplay among Li-ion transport, electrochemical reaction, and mechanical deformation. Kamikawa et al. developed a chemo-electro-mechanical phase-field model for Li penetration in LLZO, incorporating both grain-boundary conduction and interfacial voids[161]. Subsequent studies have expanded this approach to mesoscale chemo-mechanical coupling, enabling predictive simulations of dendrite nucleation and fracture propagation. In particular, phase-field frameworks incorporating ionic concentration and elastic fields have successfully reproduced dendrite growth and void- or crack-assisted failure initiation in SEs. Complementary atomistic and experimental studies further reveal dendrite-related instabilities and cracking-dominated failure in LLZO[162] and Li6PS5Cl[163].
Bridging atomistic and continuum scales, recent work has shown that MLIPs enable quantitative analysis of lithium redistribution and stress accumulation at grain boundaries, providing a consistent explanation for experimentally observed cracking and interfacial degradation during cycling[162]. The integration of atomistic and mesoscale modeling thus establishes a quantitative framework for understanding how mechanical stress and ionic transport jointly influence failure in SEs, bridging atomistic mechanisms with macroscopic reliability.
At the interfacial level, modeling of SEIs has also advanced from data-driven approaches. Because SEI formation involves nonlinear reactions and structural evolution across several length and time scales, conventional atomistic simulations often fail to capture their configurational diversity and kinetics. A property-regressed variational autoencoder trained on kinetic Monte Carlo data represents SEI configurations within a latent space organized by their physical properties, allowing the generation of structures with specified reaction barriers or ionic conductivities. This approach connects atomic-scale reaction pathways with measurable interphase performance, providing a route toward the rational design of stable, conductive interfaces in next-generation batteries[164].
From an agent's perspective, these multiscale models provide complementary decision signals rather than isolated simulations. For example, an AI agent can combine DFT-predicted interfacial reaction energies and defect formation energies with MD-derived Li-ion diffusivity and phase-field-predicted stress localization to diagnose failure modes in SEs. If phase-field simulations indicate stress concentration at grain boundaries under a given current density, while MLIP-MD reveals suppressed Li-ion mobility in the same regions, the agent can infer a mechanically driven transport bottleneck and propose design strategies such as grain-boundary chemical modification or microstructure densification. Similarly, by jointly analyzing MD-derived transport anisotropy and phase-field-predicted dendrite propagation paths, AI agents can prioritize compositions or processing routes that balance ionic conductivity and mechanical robustness. Such integration transforms multiscale modeling from descriptive analysis into actionable, agent-driven electrolyte design.
Materials ontology
In modern materials research, ontology-based frameworks provide a rigorous semantic infrastructure to formally represent data, thereby enhancing knowledge integration[165,166]. In battery and SE discovery, ontologies provide a formal, semantic framework to encode domain knowledge, allowing AI models to understand relationships between process, structure, properties, and performance[167-171]. This enables the reasoning models and AI agents to make inferences and decisions grounded in scientific domain knowledge, leveraging relationships between concepts, rather than a simple haystack of raw data. For example, inferring that two SE compositions might share a conduction mechanism if their structural descriptors are ontologically related.
The Elementary Multiperspective Material Ontology (EMMO), a multidisciplinary ontology for applied physical sciences, provides a foundation of domain ontologies by defining fundamental concepts across scales[172]. Building on this foundation, the Characterization Methodology Ontology (CHAMEO) standardizes how experimental techniques are described, ensuring uniform representation of characterization data (e.g., ionic conductivity measurements or interfacial microscopy results) across studies[173]. By mapping SE-specific entities such as ionic conductivity and electrode-electrolyte interface structure into the framework, researchers can semantically link composition, structure, and performance data.
As a concrete example, ontology-enabled AI agents can infer SE design hypotheses by reasoning over semantic relations encoded in frameworks such as EMMO. If a knowledge graph captures that high ionic conductivity in halide electrolytes correlates with anion polarizability and lattice softness, and that partial halogen substitution systematically tunes these attributes, the agent can infer that substituting Br- for Cl- in a Li3YCl6-type framework may enhance Li+ mobility while maintaining phase stability. When such compositional reasoning is combined with CHAMEO-annotated synthesis metadata, such as compatible processing temperatures and atmospheres, the agent can further constrain its inference to experimentally feasible synthesis routes. In this way, ontology-guided reasoning enables AI agents to progress from data retrieval to hypothesis generation and decision-making grounded in domain semantics.
Comparable ontology-driven inference has already been demonstrated in adjacent battery research domains. For example, the Battery Testing Ontology (BTO), aligned with EMMO and interoperable with CHAMEO, shows how semantically encoded knowledge of test procedures, hardware configurations, and measurement constraints can be leveraged by AI systems to infer consistent and executable battery testing strategies[174]. Together, these examples indicate that ontology-enabled agents can support actionable inference across both materials design and experimental decision-making in solid-state battery research.
This yields a coherent knowledge base where, for example, a microstructural feature of SE and its ionic conductivity are connected via well-defined ontological relations[170]. A high-level overview of these ontologies and their semantic layers is shown in Figure 9A and B.
Figure 9. Ontology-guided framework for SE research integrating structured knowledge, AI reasoning, and autonomous experimentation. (A) Semantic layer linking physical assets, sensors, and experiments to a shared data/model space. Reproduced with permission from ref.[167]. Copyright 2024, Wiley-VCH; (B) Multi-aspect ontology structure defining composition-structure-property-performance relations. Reproduced with permission from ref.[170]. Copyright 2022, Wiley-VCH; (C) MatKG resource description framework (RDF) schema showing semantic connections among materials-related entities, properties, bibliographic attributes, relational nodes, and external knowledge sources. Reproduced with permission from ref.[175]. Copyright 2024, Springer Nature; (D) AI agents using interoperable ontologies to drive closed-loop experimentation, hypothesis generation, and real-time knowledge updates. NER: Named entity recognition: DBpedia: a nucleus for a web of open data; SE: solid electrolyte; AI: artificial intelligence; MatKG: a knowledge graph in materials science.
In a broader context, large-scale knowledge graphs such as a knowledge graph in materials science (MatKG) demonstrate how material-science ontologies scale: materials, properties, applications, and methods, illustrating the power of ontology-based integration to uncover patterns in vast datasets[175]. The underlying schema of such ontology-driven knowledge graphs is illustrated in Figure 9C. Similarly, cross-domain frameworks such as The World Avatar demonstrate how ontologies enable interoperability beyond a single field. The World Avatar uses a dynamic knowledge graph continuously updated by autonomous agents as a digital twin of the real world[176]. Such architectures indicate that SE ontologies can interoperate on a larger scale, linking molecular-level electrolyte data to broader energy system models in a unified, machine-readable way. This illustrates the potential of ontology frameworks to accelerate materials innovation by enabling improved data integration and intelligent inference capabilities.
Ontology frameworks are also catalyzing the advent of self-driving laboratories for SE design and development by providing the semantic backbone for closed-loop experimentation[177,178]. For instance, impedance spectroscopy results or a new SE phase identified are automatically collected and annotated with rich metadata (e.g., synthesis conditions and instruments) defined by the ontology, ensuring machine-readable context. AI agents then apply automated reasoning over this structured knowledge to select the next experiments: the AI planner deduces which compositional adjustment or processing condition could improve ionic conductivity. This closed-loop feedback process continuously updates the knowledge graph with new findings, and the ontology’s formal semantics ensure that the AI’s decisions are grounded in consistent, verifiable knowledge. This AI-driven closed-loop workflow, enabled by interoperable ontologies, is conceptually summarized in Figure 9D. By coupling ontology-driven information management with automated experimentation, self-driving labs can accelerate SE discovery and achieve rapid optimization through an intelligent framework.
FUTURE PERSPECTIVES
AI has significantly accelerated the discovery and design of SEs, yet several challenges continue to limit its widespread adoption. The most fundamental issue lies in data quality and coverage. While Li-based systems are well represented, datasets for multivalent ion conductors such as Mg2+, Ca2+, and Al3+ remain scarce and inconsistent. These gaps undermine model transferability and reliability, highlighting the need for standardized, openly accessible databases to sustain AI-guided discovery[179]. A second challenge is interpretability. Deep learning models often act as black boxes, offering predictions without a physical rationale. Explainable AI is beginning to resolve this issue. For example, Explainable ElemNet (XelemNet) embeds interpretability tools within the ElemNet (a deep-learning model trained using elemental composition) framework and reveals that predicted formation energies and stability trends align with chemical intuition even without explicit periodic descriptors[180].
Figure 10 summarizes representative applications of ML and AI in solid-state battery research. The transformer-based crystal graph network transformer (CGformer) model [Figure 10A] captures long-range correlations and efficiently screens vast compositional spaces, identifying high-conductivity NASICON-type structures[181]. ML coupled with phase field simulations [Figure 10B] clarifies how stack pressure and current density influence void evolution in LLZO and argyrodite systems[182]. At the anode, support vector and kernel ridge models identify dopants such as Sc3+ and Ca2+ that stabilize Li/LLZO interfaces [Figure 10C][183]. At the cathode, image-based learning automates microstructural analysis and quantifies interfacial morphology [Figure 10D][184].
Figure 10. Representative physical and computational models for solid-state battery design. (A) Transformer-based CGformer architecture for screening high-entropy SEs. Reproduced with permission from ref.[181]. Copyright 2025, Cell Press; (B) Machine-learning-assisted phase-field modeling of void-evolution modes in LLZO systems. Reproduced with permission from ref.[182], Copyright 2025, Wiley-VCH; (C) Screening of cation-doped LLZO compositions thermodynamically stable against Li metal using ML models. Reproduced with permission from ref.[183]. Copyright 2019, Royal Society of Chemistry; (D) Image-based workflow for cathode microstructure quantification, including phase segmentation, feature extraction, and line-intercept analysis. Reproduced with permission from ref.[184]. Copyright 2025, Wiley-VCH; (E) Evolution of research paradigms from knowledge-augmented human investigation to autonomous AI-agent-driven discovery. DFT: density functional theory; SE: solid electrolyte; LLZO: Li7La3Zr2O12; ML: machine learning; AI: artificial intelligence.
Collectively, these studies illustrate a transition from empirical optimization to knowledge-driven discovery. AI should function as an integrated design engine linking theory, computation, and experiment within unified frameworks[181]. As AI systems evolve from predictive models toward autonomous agents, the central challenge shifts from what AI can optimize to how agent decisions should be guided, validated, and trusted in practice. Human-in-the-loop AI agents play a critical role in this transition by enabling real-time interaction between automated reasoning and scientific judgment. Rather than operating as fully autonomous black boxes, emerging frameworks allow researchers to impose constraints, refine objectives, validate intermediate hypotheses, and override unphysical or unsafe actions. Recent hybrid human-AI systems demonstrate that expert feedback can effectively steer agent exploration toward physically meaningful regions of the design space, while agents handle large-scale search, data integration, and uncertainty-aware decision prioritization. Such collaboration is particularly important for SEs, where safety, interfacial stability, and manufacturability constraints must be continuously assessed. Human-in-the-loop paradigms, therefore, provide a practical pathway for ensuring interpretability, experimental safety, and scientific accountability as AI agents move toward higher levels of autonomy.
Proof of concept systems already support this vision: multi-tool scientific agents such as Scientific tool agent (SciToolAgent) coordinate distributed computational resources through knowledge-graph architectures[185], while combined LLM-human workflows based on GPT-4 have been used to propose and evaluate candidate electrolytes for Zn-I2 batteries[186].
Figure 10E highlights the ongoing evolution from human-centered research toward autonomous discovery. In the early stages, LLMs assist researchers in literature mining and property prediction; at advanced stages, AI agents integrate theory, synthesis, and testing within continuous feedback loops. Knowledge becomes cumulative and reusable, enabling adaptive and self-improving materials discovery.
Realizing autonomous, agent-based workflows for SE ultimately requires rigorous FAIR data stewardship. Beyond findability and accessibility, interoperability remains the critical bottleneck. Ontology gaps persist in (i) synthesis parameters (e.g., temperature, pressure, precursor ratios, annealing conditions) and (ii) disorder metrics (e.g., defect concentration, amorphization degree, intergranular heterogeneity). The lack of standardized, machine-readable vocabularies and provenance schemas hinders knowledge transfer across laboratories, databases, and modalities. Community efforts are needed to (1) extend existing materials ontologies with SE-specific synthesis and disorder descriptors; (2) adopt minimal metadata checklists (instrument type, atmosphere, pellet density, stack pressure, impedance geometry, temperature ramp/hold) in electronic lab notebooks and repositories; and (3) enforce versioned provenance and uncertainty annotations so that agent decisions are transparent and auditable. Embedding these elements into FAIR-compliant infrastructures will enable cross-lab agent learning and establish a foundation for closed-loop discovery that is traceable, reproducible, and physically grounded.
In addition to data and ontology challenges, translating laboratory-scale SE performance into industrially relevant outcomes remains a major barrier. Many high-performing candidates identified through computation or small-scale experiments encounter difficulties during scale-up due to processing constraints, sensitivity to moisture or impurities, precursor cost and availability, and the complexity of interfacial engineering in large-format cells. Consequently, laboratory performance does not always translate directly into industrial manufacturability or device-level stability. Incorporating metrics related to synthesizability, processability, and scalability into future AI-driven and agent-based design frameworks will therefore be essential for completing the closed-loop discovery cycle and ensuring that proposed SEs are not only high-performing but also industrially viable.
Looking ahead, five strategic directions will guide future progress:
(1) Data infrastructure: establishing FAIR-compliant repositories from quantum to device scales, with rigorous data curation, validation, and provenance tracking to ensure reliability, transferability, and physical interpretability of models, as exemplified by databases such as DDSE.
(2) Algorithmic innovation: integrating domain knowledge with data efficiency through small sample learning, uncertainty-aware modeling, and physics-informed graph networks.
(3) Computational and experimental integration: coupling predictive modeling with robotic synthesis and operando characterization to close the design-test-learn cycle.
(4) Beyond incremental discovery: developing agents capable of decision-aware multi-objective optimization across conductivity, stability, and synthesizability to explore new SE frameworks.
(5) Global consortium for data and agent: fostering international collaboration to build open, interoperable platforms that connect data, algorithms, and agents, accelerating innovation in the global community.
In summary, the convergence of interpretable algorithms, theoretical modeling, and autonomous experimentation marks a paradigm shift in SE research. AI is expected to evolve from a predictive assistant to an active collaborator, accelerating optimization today and enabling self-correcting materials discovery in the future.
DECLARATIONS
Acknowledgments
The authors acknowledge the resources and administrative support provided by the Core Research Cluster for Materials Science (CRC-MS) and the Advanced Institute for Materials Research (WPI-AIMR), Tohoku University.
Authors’ contributions
Investigation: Wang, Q.
Formal analysis: Wang, Q.; Sato, R.; García-Méndez, R.; Ou, P.; Soon, A.; Zhao, J.; Wang, X.; Orimo, S.; Jang, W.
Writing - original draft: Wang, Q.; Jang, W.
Writing - review & editing: Wang, Q.; Sato, R.; García-Méndez, R.; Ou, P.; Soon, A.; Zhao, J.; Wang, X.; Orimo, S.; Cheng, E. J.
Validation: Sato, R.; García-Méndez, R.; Ou, P.; Soon, A.; Zhao, J.; Wang, X.; Orimo, S.; Cheng, E. J.
Supervision: Cheng, E. J.
Resources: Cheng, E. J.
Funding acquisition: Cheng, E. J.
Project administration: Cheng, E. J.
Availability of data and materials
Not applicable.
Financial support and sponsorship
Wang, Q. and Cheng, E. J. acknowledge financial support from the GIMRT Program of the Institute for Materials Research (IMR), Tohoku University (Proposal No. 202412-CRKEQ-0206), the AY2024/2025 TUMUG Support Program, and the Basic Research Grant from the TEPCO Memorial Foundation. Zhao, J. acknowledges support from the National Natural Science Foundation of China (Grant Nos. 22561142235 and 52471223) and the Science and Technology Commission of Shanghai Municipality (Grant No. 23160714000).
Conflicts of interest
Soon, A. is an Associate Editor of the journal AI Agent. Wang, X. and Cheng, E. J. serve as Editorial Board Members of AI Agent. They were not involved in any steps of the editorial process, including reviewer selection, manuscript handling, or decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Dutra, A. C. C.; Goldmann, B. A.; Islam, M. S.; Dawson, J. A. Understanding solid-state battery electrolytes using atomistic modelling and machine learning. Nat. Rev. Mater. 2025, 10, 566-83.
2. Chen, L.; Msigwa, G.; Yang, M.; et al. Strategies to achieve a carbon neutral society: a review. Environ. Chem. Lett. 2022, 20, 2277-310.
3. Kalnaus, S.; Dudney, N. J.; Westover, A. S.; Herbert, E.; Hackney, S. Solid-state batteries: The critical role of mechanics. Science 2023, 381, eabg5998.
4. Li, Y.; Song, S.; Kim, H.; et al. A lithium superionic conductor for millimeter-thick battery electrode. Science 2023, 381, 50-3.
5. Janek, J.; Zeier, W. G. Challenges in speeding up solid-state battery development. Nat. Energy. 2023, 8, 230-40.
6. Kamaya, N.; Homma, K.; Yamakawa, Y.; et al. A lithium superionic conductor. Nat. Mater. 2011, 10, 682-6.
7. Xiao, Y.; Wang, Y.; Bo, S.; Kim, J. C.; Miara, L. J.; Ceder, G. Understanding interface stability in solid-state batteries. Nat. Rev. Mater. 2019, 5, 105-26.
8. Kwak, H.; Kim, J. S.; Han, D.; et al. Boosting the interfacial superionic conduction of halide solid electrolytes for all-solid-state batteries. Nat. Commun. 2023, 14, 2459.
9. Cheng, E. J.; Yang, T.; Liu, Y.; et al. Correlation between mechanical properties and ionic conductivity of polycrystalline sodium superionic conductors: a relative density-dominant relationship. Mater. Today. Energy. 2024, 44, 101644.
11. Urban, A.; Seo, D.; Ceder, G. Computational understanding of Li-ion batteries. NPJ. Comput. Mater. 2016, 2, 16002.
12. Murugan, R.; Thangadurai, V.; Weppner, W. Fast lithium ion conduction in garnet-type Li7La3Zr2O12. Angew. Chem. Int. Ed. Engl. 2007, 46, 7778-81.
13. Zhao, Y.; Daemen, L. L. Superionic conductivity in lithium-rich anti-perovskites. J. Am. Chem. Soc. 2012, 134, 15042-7.
14. Asano, T.; Sakai, A.; Ouchi, S.; Sakaida, M.; Miyazaki, A.; Hasegawa, S. Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 2018, 30, e1803075.
15. Hirose, T.; Matsui, N.; Itoh, T.; et al. High-capacity, reversible hydrogen storage using H--conducting solid electrolytes. Science 2025, 389, 1252-5.
16. Zhao, F.; Zhang, S.; Wang, S.; et al. Anion sublattice design enables superionic conductivity in crystalline oxyhalides. Science 2025, 390, 199-204.
17. Aykol, M.; Herring, P.; Anapolsky, A. Machine learning for continuous innovation in battery technologies. Nat. Rev. Mater. 2020, 5, 725-7.
18. Lombardo, T.; Duquesnoy, M.; El-Bouysidy, H.; et al. Artificial intelligence applied to battery research: hype or reality? Chem. Rev. 2022, 122, 10899-969.
19. Huang, S.; Cole, J. M. BatteryDataExtractor: battery-aware text-mining software embedded with BERT models. Chem. Sci. 2022, 13, 11487-95.
20. Dunn, A.; Wang, Q.; Ganose, A.; Dopp, D.; Jain, A. Benchmarking materials property prediction methods: the Matbench test set and Automatminer reference algorithm. NPJ. Comput. Mater. 2020, 6, 138.
21. Chen, H.; Liu, H.; Tew, Y.; Ren, X.; Tang, X.; Wang, X. Distilling knowledge from catalysis literature with long-context large language model agents. ACS. Catal. 2025, 15, 18244-54.
22. Goodenough, J. B.; Park, K. S. The Li-ion rechargeable battery: a perspective. J. Am. Chem. Soc. 2013, 135, 1167-76.
23. Butler, K. T.; Davies, D. W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547-55.
24. Jun, K.; Chen, Y.; Wei, G.; Yang, X.; Ceder, G. Diffusion mechanisms of fast lithium-ion conductors. Nat. Rev. Mater. 2024, 9, 887-905.
25. Sau, K.; Takagi, S.; Ikeshoji, T.; et al. Unlocking the secrets of ideal fast ion conductors for all-solid-state batteries. Commun. Mater. 2024, 5, 122.
26. Mo, Y.; Ong, S. P.; Ceder, G. First Principles Study of the Li10GeP2S12 lithium super ionic conductor material. Chem. Mater. 2011, 24, 15-7.
27. Sendek, A. D.; Yang, Q.; Cubuk, E. D.; Duerloo, K. N.; Cui, Y.; Reed, E. J. Holistic computational structure screening of more than 12,000 candidates for solid lithium-ion conductor materials. Energy. Environ. Sci. 2017, 10, 306-20.
28. Ahmad, Z.; Xie, T.; Maheshwari, C.; Grossman, J. C.; Viswanathan, V. Machine learning enabled computational screening of inorganic solid electrolytes for suppression of dendrite formation in lithium metal anodes. ACS. Cent. Sci. 2018, 4, 996-1006.
29. Wang, Y.; Xie, T.; France-lanord, A.; et al. Toward designing highly conductive polymer electrolytes by machine learning assisted coarse-grained molecular dynamics. Chem. Mater. 2020, 32, 4144-51.
30. Kahle, L.; Marcolongo, A.; Marzari, N. High-throughput computational screening for solid-state Li-ion conductors. Energy. Environ. Sci. 2020, 13, 928-48.
31. Funke, K. Solid state ionics: from Michael Faraday to green energy-the European dimension. Sci. Technol. Adv. Mater. 2013, 14, 043502.
32. Nernst, W.; Wild, W. Einiges über das Verhalten elektrolytischer Glühkörper. Zeitschrift. für. Elektrochemie. 2010, 7, 373-6. (in German).
33. Tubandt, C.; Lorenz, E. The molecular condition and electrical conductivity of crystallized salts. Z. Phys. Chem. 1914, 87, 513-42. (in German).
34. Wagner, C.; Schottky, W. Theorie der geordneten Mischphasen. Z. Phys. Chem. B. 1930, 163-210. (in German).
35. Yao, Y. F. Y.; Kummer, J. T. Ion exchange properties of and rates of ionic diffusion in beta-alumina. J. Inorg. Nucl. Chem. 1967, 29, 2453-75.
36. Takahashi, T.; Yamamoto, O.; Yamada, S.; Hayashi, S. Solid-state ionics: high copper ion conductivity of the system CuCl-CuI-RbCl. J. Electrochem. Soc. 2019, 126, 1654-8.
37. Goodenough, J.; Hong, H.; Kafalas, J. Fast Na+-ion transport in skeleton structures. Mater. Res. Bull. 1976, 11, 203-20.
38. Hong, H. Crystal structure and ionic conductivity of Li14Zn(GeO4)4 and other new Li+ superionic conductors. Mater. Res. Bull. 1978, 13, 117-24.
40. Jansen, M.; Henseler, U. Synthesis, structure determination, and ionic conductivity of sodium tetrathiophosphate. J. Solid. State. Chem. 1992, 99, 110-9.
41. Kanno, R.; Hata, T.; Kawamoto, Y.; Irie, M. Synthesis of a new lithium ionic conductor, thio-LISICON-lithium germanium sulfide system. Solid. State. Ionics. 2000, 130, 97-104.
42. Inaguma, Y.; Liquan, C.; Itoh, M.; et al. High ionic conductivity in lithium lanthanum titanate. Solid. State. Commun. 1993, 86, 689-93.
43. Chen, C. Ionic conductivity, lithium insertion and extraction of lanthanum lithium titanate. Solid. State. Ionics. 2001, 144, 51-7.
44. Thangadurai, V.; Kaack, H.; Weppner, W. J. F. Novel fast lithium ion conduction in garnet-type Li5La3M2O12 (M = Nb, Ta). J. Am. Ceram. Soc. 2004, 86, 437-40.
45. Verbraeken, M. C.; Suard, E.; Irvine, J. T. S. Structural and electrical properties of calcium and strontium hydrides. J. Mater. Chem. 2009, 19, 2766.
46. Verbraeken, M. C.; Cheung, C.; Suard, E.; Irvine, J. T. High H- ionic conductivity in barium hydride. Nat. Mater. 2015, 14, 95-100.
47. Kobayashi, G.; Hinuma, Y.; Matsuoka, S.; et al. Pure H- conduction in oxyhydrides. Science 2016, 351, 1314-7.
48. Takeiri, F.; Watanabe, A.; Okamoto, K.; et al. Hydride-ion-conducting K2NiF4-type Ba-Li oxyhydride solid electrolyte. Nat. Mater. 2022, 21, 325-30.
49. Zhang, W.; Cui, J.; Wang, S.; et al. Deforming lanthanum trihydride for superionic conduction. Nature 2023, 616, 73-6.
50. Cui, J.; Zou, R.; Zhang, W.; et al. A room temperature rechargeable all-solid-state hydride ion battery. Nature 2025, 646, 338-42.
51. Cheng, E. J.; Duan, H.; Wang, M. J.; et al. Li-stuffed garnet solid electrolytes: current status, challenges, and perspectives for practical Li-metal batteries. Energy. Storage. Mater. 2025, 75, 103970.
52. Schmidt, J.; Marques, M. R. G.; Botti, S.; Marques, M. A. L. Recent advances and applications of machine learning in solid-state materials science. NPJ. Comput. Mater. 2019, 5, 83.
53. Rashid, A. B.; Kausik, M. A. K. AI revolutionizing industries worldwide: a comprehensive overview of its diverse applications. Hybrid. Adv. 2024, 7, 100277.
54. Cheng, E. J.; Sharafi, A.; Sakamoto, J. Intergranular Li metal propagation through polycrystalline Li6.25Al0.25La3Zr2O12 ceramic electrolyte. Electrochim. Acta. 2017, 223, 85-91.
55. Wu, J.; Liu, S.; Han, F.; Yao, X.; Wang, C. Lithium/sulfide all-solid-state batteries using sulfide electrolytes. Adv. Mater. 2021, 33, e2000751.
56. Richards, W. D.; Miara, L. J.; Wang, Y.; Kim, J. C.; Ceder, G. Interface stability in solid-state batteries. Chem. Mater. 2015, 28, 266-73.
57. He, B.; Zhang, F.; Xin, Y.; et al. Halogen chemistry of solid electrolytes in all-solid-state batteries. Nat. Rev. Chem. 2023, 7, 826-42.
58. Wang, Q.; Li, H.; Zhang, R.; et al. Oxygen vacancies boosted fast Mg2+ migration in solids at room temperature. Energy. Storage. Mater. 2022, 51, 630-7.
59. Yin, J.; Xu, X.; Jiang, S.; et al. High ionic conductivity PEO-based electrolyte with 3D framework for dendrite-free solid-state lithium metal batteries at ambient temperature. Chem. Eng. J. 2022, 431, 133352.
60. Li, R.; Hua, H.; Zeng, Y.; et al. Promote the conductivity of solid polymer electrolyte at room temperature by constructing a dual range ionic conduction path. J. Energy. Chem. 2022, 64, 395-403.
61. Gong, S.; Zhang, Y.; Mu, Z.; et al. A predictive machine learning force-field framework for liquid electrolyte development. Nat. Mach. Intell. 2025, 7, 543-52.
62. Dave, A.; Mitchell, J.; Burke, S.; Lin, H.; Whitacre, J.; Viswanathan, V. Autonomous optimization of non-aqueous Li-ion battery electrolytes via robotic experimentation and machine learning coupling. Nat. Commun. 2022, 13, 5454.
63. Flores, E.; Wölke, C.; Yan, P.; et al. Learning the laws of lithium-ion transport in electrolytes using symbolic regression. Digit. Discov. 2022, 1, 440-7.
64. He, X.; Zhu, Y.; Mo, Y. Origin of fast ion diffusion in super-ionic conductors. Nat. Commun. 2017, 8, 15893.
65. Gautam, A.; Sadowski, M.; Ghidiu, M.; et al. Engineering the site-disorder and lithium distribution in the lithium superionic argyrodite Li6PS5Br. Adv. Energy. Mater. 2020, 11, 2003369.
66. Schwietert, T. K.; Vasileiadis, A.; Wagemaker, M. First-principles prediction of the electrochemical stability and reaction mechanisms of solid-state electrolytes. JACS. Au. 2021, 1, 1488-96.
67. López, C.; Rurali, R.; Cazorla, C. How concerted are ionic hops in inorganic solid-state electrolytes? J. Am. Chem. Soc. 2024, 146, 8269-79.
68. Sato, R.; Ando, Y.; Sau, K.; Shibuta, Y. Visualizing concerted ion migration of superionic conductors via directed graphs. Chem. Mater. 2025.
69. Xu, H.; Cao, G.; Shen, Y.; et al. Enabling argyrodite sulfides as superb solid-state electrolyte with remarkable interfacial stability against electrodes. Energy. &. Environ. Mater. 2022, 5, 852-64.
70. Morgan, B. J. Mechanistic origin of superionic lithium diffusion in anion-disordered Li6PS5X argyrodites. Chem. Mater. 2021, 33, 2004-18.
71. Golov, A.; Carrasco, J. Molecular-level insight into the interfacial reactivity and ionic conductivity of a Li-argyrodite Li6PS5Cl solid electrolyte at bare and coated Li-metal anodes. ACS. Appl. Mater. Interfaces. 2021, 13, 43734-45.
72. Kato, Y.; Hori, S.; Saito, T.; et al. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy. 2016, 1, 16030.
73. Hou, J.; Sun, W.; Yuan, Q.; et al. Multiscale engineered bionic solid-state electrolytes breaking the stiffness-damping trade-off. Angew. Chem. Int. Ed. Engl. 2025, 64, e202421427.
74. Baba, T.; Kajita, S.; Shiga, T.; Ohba, N. Fast evaluation technique for the shear viscosity and ionic conductivity of electrolyte solutions. Sci. Rep. 2022, 12, 7291.
75. Fujimura, K.; Seko, A.; Koyama, Y.; et al. Accelerated materials design of lithium superionic conductors based on first-principles calculations and machine learning algorithms. Adv. Energy. Mater. 2013, 3, 980-5.
76. Wang, Z.; Lin, X.; Han, Y.; et al. Harnessing artificial intelligence to holistic design and identification for solid electrolytes. Nano. Energy. 2021, 89, 106337.
77. Lao, Z.; Tao, K.; Xiao, X.; et al. Data-driven exploration of weak coordination microenvironment in solid-state electrolyte for safe and energy-dense batteries. Nat. Commun. 2025, 16, 1075.
78. Hargreaves, C. J.; Gaultois, M. W.; Daniels, L. M.; et al. A database of experimentally measured lithium solid electrolyte conductivities evaluated with machine learning. NPJ. Comput. Mater. 2023, 9, 9.
79. Lin, S.; Lin, Y.; He, B.; et al. Reclaiming neglected compounds as promising solid state electrolytes by predicting electrochemical stability window with dynamically determined decomposition pathway. Adv. Energy. Mater. 2022, 12, 2201808.
80. Chen, J.; Jiang, L.; Tan, S.; et al. Machine-learning-aided screening of inorganic lithium solid-state electrolytes with a wide electrochemical window. J. Mater. Chem. A. 2025, 13, 23445-53.
81. Zhang, Y.; He, X.; Chen, Z.; et al. Unsupervised discovery of solid-state lithium ion conductors. Nat. Commun. 2019, 10, 5260.
82. Lee, B. D.; Lee, J. W.; Park, J.; Cho, M. Y.; Park, W. B.; Sohn, K. S. Argyrodite configuration determination for DFT and AIMD calculations using an integrated optimization strategy. RSC. Adv. 2022, 12, 31156-66.
83. Bai, C.; Li, Y.; Xiao, G.; et al. Understanding the electrochemical window of solid-state electrolyte in full battery application. Chem. Rev. 2025, 125, 6541-608.
84. Wang, S.; Gong, S.; Böger, T.; et al. Multimodal machine learning for materials science: discovery of novel li-ion solid electrolytes. Chem. Mater. 2024, 36, 11541-50.
85. Kim, J.; Lee, D.; Lee, D.; Li, X.; Lee, Y. L.; Kim, S. Machine learning prediction models for solid electrolytes based on lattice dynamics properties. J. Phys. Chem. Lett. 2024, 15, 5914-22.
86. Cortés, H. A.; Bonilla, M. R.; Früchtl, H.; Van Mourik, T.; Carrasco, J.; Akhmatskaya, E. A data-mining approach to understanding the impact of multi-doping on the ionic transport mechanism of solid electrolytes materials: the case of dual-doped Ga0.15/ScyLi7La3Zr2O12. J. Mater. Chem. A. 2024, 12, 5181-93.
87. Shen, Z.; Bao, W.; Dang, Z.; et al. Predicting the ionic conductivity and obtaining mechanistic insights of plasticized solid polymer electrolytes using a data-driven approach. Macromolecules 2025, 58, 4194-205.
88. Zhang, Z.; Chu, J.; Zhang, H.; Liu, X.; He, M. Mining ionic conductivity descriptors of antiperovskite electrolytes for all-solid-state batteries via machine learning. J. Energy. Storage. 2024, 75, 109714.
89. Zhao, Q.; Zhang, L.; He, B.; et al. Identifying descriptors for Li+ conduction in cubic Li-argyrodites via hierarchically encoding crystal structure and inferring causality. Energy. Storage. Mater. 2021, 40, 386-93.
90. Chaudhary, A. K. Development of highly dense Ga-LLZO solid electrolyte pallet for all-solid-state battery using machine learning approach. Solid. State. Commun. 2025, 404, 116038.
91. Liu, M.; Clement, C.; Liu, K.; Wang, X.; Sparks, T. D. A data science approach for advanced solid polymer electrolyte design. Comput. Mater. Sci. 2021, 187, 110108.
92. Bradford, G.; Lopez, J.; Ruza, J.; et al. Chemistry-informed machine learning for polymer electrolyte discovery. ACS. Cent. Sci. 2023, 9, 206-16.
93. Lomeli, E. G.; Ransom, B.; Ramdas, A.; et al. Predicting reactivity and passivation of solid-state battery interfaces. ACS. Appl. Mater. Interfaces. 2024, 16, 51584-94.
94. Mortazavi, B. Recent advances in machine learning-assisted multiscale design of energy materials. Adv. Energy. Mater. 2024, 15, 2403876.
95. Ko, T. W.; Ong, S. P. Recent advances and outstanding challenges for machine learning interatomic potentials. Nat. Comput. Sci. 2023, 3, 998-1000.
96. You, Y.; Zhang, D.; Wu, F.; et al. Principal component analysis enables the design of deep learning potential precisely capturing LLZO phase transitions. NPJ. Comput. Mater. 2024, 10, 57.
97. Lee, J.; Ju, S.; Hwang, S.; et al. Disorder-dependent Li diffusion in Li6PS5Cl investigated by machine-learning potential. ACS. Appl. Mater. Interfaces. 2024, 16, 46442-53.
98. Guo, X.; Wang, Z.; Yang, J.; Gong, X. Machine-learning assisted high-throughput discovery of solid-state electrolytes for Li-ion batteries. J. Mater. Chem. A. 2024, 12, 10124-36.
99. Lian, J.; Fu, X.; Gong, X.; Xiao, R.; Li, H. High-throughput NEB for Li-ion conductor discoveryvia fine-tuned CHGNet potential. J. Mater. Chem. A. 2025, 13, 34918-26.
100. Dembitskiy, A. D.; Humonen, I. S.; Eremin, R. A.; Aksyonov, D. A.; Fedotov, S. S.; Budennyy, S. A. Benchmarking machine learning models for predicting lithium ion migration. NPJ. Comput. Mater. 2025, 11, 131.
101. Du, H.; Huang, X.; Hui, J.; Zhang, L.; Zhou, Y.; Wang, H. Assessment and application of universal machine learning interatomic potentials in solid-state electrolyte research. ACS. Materials. Lett. 2025, 7, 3403-12.
102. Deng, Y.; Li, Y.; Sai Gautam, G.; Zhu, B.; Deng, Z. Accelerating the discovery of disordered multi-component solid-state electrolytes using machine learning interatomic potentials. J. Mater. Chem. A. 2025, 13, 34507-18.
103. Batzner, S.; Musaelian, A.; Sun, L.; et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 2022, 13, 2453.
104. Zhang, L.; Han, J.; Wang, H.; Car, R.; E, W. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 2018, 120, 143001.
105. Yao, K.; Herr, J. E.; Toth, D. W.; Mckintyre, R.; Parkhill, J. The TensorMol-0.1 model chemistry: a neural network augmented with long-range physics. Chem. Sci. 2018, 9, 2261-9.
106. Jain, A.; Ong, S. P.; Hautier, G.; et al. Commentary: The materials project: a materials genome approach to accelerating materials innovation. APL. Materials. 2013, 1, 011002.
107. Deng, B.; Zhong, P.; Jun, K.; et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 2023, 5, 1031-41.
108. Chen, C.; Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2022, 2, 718-28.
109. Hwang, W.; Victor Oh, S. H.; Shin, J.; Soon, A.; Yoo, S. H.; Jang, W. Data-driven materials informatics for novel piezoelectric Janus-type nanomaterials discovery. J. Phys. Chem. Lett. 2024, 15, 6451-7.
110. Batatia, I.; Benner, P.; Chiang, Y.; et al. A foundation model for atomistic materials chemistry. J. Chem. Phys. 2025, 163, 184110.
111. Alghamdi, N.; de Angelis, P.; Asinari, P.; et al. Comparing fine-tuning strategies of MACE machine learning force field for modeling Li-ion diffusion in LiF for batteries.
112. Böhm, J.; Champagne, A. Predicting crystal structures and ionic conductivity in Li3YCl6-xBrx halide solid electrolytes using a fine-tuned machine learning interatomic potential.
113. Kim, J.; Lee, J.; Oh, S.; et al. An efficient forgetting-aware fine-tuning framework for pretrained universal machine-learning interatomic potentials.
114. Deng, B.; Choi, Y. Zhong. P.; et al. Overcoming systematic softening in universal machine learning interatomic potentials by fine-tuning.
115. Liu, X.; Zeng, K.; Luo, Z.; et al. Fine-tuning universal machine-learned interatomic potentials: a tutorial on methods and applications.
116. Chen, Z.; Du, T.; Krishnan, N. M. A.; Yue, Y.; Smedskjaer, M. M. Disorder-induced enhancement of lithium-ion transport in solid-state electrolytes. Nat. Commun. 2025, 16, 1057.
117. Wang, Q.; Yang, F.; Wang, Y.; et al. Unraveling the complexity of divalent hydride electrolytes in solid-state batteries via a data-driven framework with large language model. Angew. Chem. Int. Ed. Engl. 2025, 64, e202506573.
118. Leng, Y.; Zhong, Y.; Gu, Z.; et al. Intelligent, personalized scientific assistant via large language models for solid-state battery research. ACS. Mater. Lett. 2025, 7, 1807-16.
119. Chen, X.; Liu, M.; Yin, S.; Gao, Y. C.; Yao, N.; Zhang, Q. Uni-electrolyte: an artificial intelligence platform for designing electrolyte molecules for rechargeable batteries. Angew. Chem. Int. Ed. Engl. 2025, 64, e202503105.
120. Zhang, D.; Jia, X.; Hung, T. B.; et al. “DIVE” into hydrogen storage materials discovery with AI agents.
121. Gomes, C. P.; Bai, J.; Xue, Y.; et al. CRYSTAL: a multi-agent AI system for automated mapping of materials’ crystal structures. MRS. Communications. 2019, 9, 600-8.
122. M Bran, A.; Cox, S.; Schilter, O.; Baldassari, C.; White, A. D.; Schwaller, P. Augmenting large language models with chemistry tools. Nat. Mach. Intell. 2024, 6, 525-35.
123. Darvish, K.; Skreta, M.; Zhao, Y.; et al. ORGANA: A robotic assistant for automated chemistry experimentation and characterization. Matter 2025, 8, 101897.
124. Szymanski, N. J.; Rendy, B.; Fei, Y.; et al. An autonomous laboratory for the accelerated synthesis of novel materials. Nature 2023, 624, 86-91.
125. Mahbub, R.; Huang, K.; Jensen, Z.; Hood, Z. D.; Rupp, J. L.; Olivetti, E. A. Text mining for processing conditions of solid-state battery electrolytes. Electrochem. Commun. 2020, 121, 106860.
126. Shon, Y. J.; Min, K. Extracting chemical information from scientific literature using text mining: building an ionic conductivity database for solid-state electrolytes. ACS. Omega. 2023, 8, 18122-7.
127. Heo, G.; Soon, A.; Lee, T. Data-mining fluoride-based solid-state electrolytes for monovalent metal batteries. J. Mater. Chem. A. 2024, 12, 27409-20.
128. Jang, S. H.; Tateyama, Y.; Jalem, R. High-throughput data-driven prediction of stable high-performance Na-ion sulfide solid electrolytes. Adv. Funct. Mater. 2022, 32, 2206036.
129. Lee, B. D.; Gavali, D. S.; Kim, H.; et al. Discovering virtual Na-based argyrodites as solid-state electrolytes using DFT, AIMD, and machine learning techniques. J. Mater. Chem. A. 2025, 13, 10462-74.
130. Chen, Y.; Liu, Y.; He, Z.; et al. Artificial intelligence for the understanding of electrolyte chemistry and electrode interface in lithium battery. NSO. 2023, 20230039.
131. Jiang, X.; Wang, W.; Tian, S.; Wang, H.; Lookman, T.; Su, Y. Applications of natural language processing and large language models in materials discovery. npj. Comput. Mater. 2025, 11, 79.
132. Zhang, J.; Chen, X.; Ye, X.; Yang, Y.; Ai, B. Large language model in materials science: roles, challenges, and strategic outlook. adv. intell. discov. 2025, 202500085.
133. Maffettone, P. M.; Banko, L.; Cui, P.; et al. Crystallography companion agent for high-throughput materials discovery. Nat. Comput. Sci. 2021, 1, 290-7.
134. Wong, L. L.; Phuah, K. C.; Dai, R.; Chen, H.; Chew, W. S.; Adams, S. Bond valence pathway analyzer - an automatic rapid screening tool for fast ion conductors within softBV. Chem. Mater. 2021, 33, 625-41.
135. Chen, H.; Wong, L. L.; Adams, S. SoftBV - a software tool for screening the materials genome of inorganic fast ion conductors. Acta. Crystallogr. B. Struct. Sci. Cryst. Eng. Mater. 2019, 75, 18-33.
136. Park, D.; Park, H.; Lee, Y.; et al. Theoretical design of lithium chloride superionic conductors for all-solid-state high-voltage lithium-ion batteries. ACS. Appl. Mater. Interfaces. 2020, 12, 34806-14.
137. Li, Z.; Dong, H.; Zhang, B. Motif-based exploration of halide classes of Li5M10.5M20.5X8 conductors using the DFT method: toward high Li-Ion conductivity and improved stability. ACS. Appl. Mater. Interfaces. 2023, 15, 42481-9.
138. Choi, E.; Jo, J.; Kim, W.; Min, K. Searching for mechanically superior solid-state electrolytes in Li-ion batteries via data-driven approaches. ACS. Appl. Mater. Interfaces. 2021, 13, 42590-7.
139. Yang, F.; Sato, R.; Cheng, E. J.; et al. Data-driven viewpoint for developing next-generation Mg-ion solid-state electrolytes. J. Electrochem. 2024, 30, 2415001.
140. Yang, F.; Campos Dos Santos, E.; Jia, X.; et al. A dynamic database of solid-state electrolyte (DDSE) picturing all-solid-state batteries. Nano. Mater. Sci. 2024, 6, 256-62.
141. Levin, I. NIST Inorganic Crystal Structure Database (ICSD), National Institute of Standards and Technology. 2018.
142. Merchant, A.; Batzner, S.; Schoenholz, S. S.; Aykol, M.; Cheon, G.; Cubuk, E. D. Scaling deep learning for materials discovery. Nature 2023, 624, 80-5.
143. Chen, C.; Nguyen, D. T.; Lee, S. J.; et al. Accelerating computational materials discovery with machine learning and cloud high-performance computing: from large-scale screening to experimental validation. J. Am. Chem. Soc. 2024, 146, 20009-18.
144. Sendek, A. D.; Cheon, G.; Pasta, M.; Reed, E. J. Quantifying the search for solid Li-ion electrolyte materials by anion: a data-driven perspective. J. Phys. Chem. C. 2020, 124, 8067-79.
145. Zhang, J.; Li, J.; Zhao, G.; Wang, Q.; Guo, Y. G.; Yang, C. Mining solid-state electrolytes from metal-organic framework databases through large language models and representation clustering. J. Am. Chem. Soc. 2025, 147, 40496-506.
146. Nguyen, T. M.; Tawfik, S. A.; Tran, T.; Gupta, S.; Rana, S.; Venkatesh, S. The search for superionic solid-state electrolytes using a physics-informed generative model. Mater. Horiz. 2025, 12, 6945-55.
147. Leong, S. X.; Griesbach, C. E.; Zhang, R.; et al. Steering towards safe self-driving laboratories. Nat. Rev. Chem. 2025, 9, 707-22.
148. Nair, M. R.; Roy, T. Role of artificial intelligence in the design and discovery of next-generation battery electrolytes. Chem. Phys. Rev. 2025, 6, 011311.
149. Noh, J.; Doan, H. A.; Job, H.; et al. An integrated high-throughput robotic platform and active learning approach for accelerated discovery of optimal electrolyte formulations. Nat. Commun. 2024, 15, 2757.
150. Wang, C.; Kim, Y. J.; Vriza, A.; et al. Autonomous platform for solution processing of electronic polymers. Nat. Commun. 2025, 16, 1498.
151. Lookman, T.; Balachandran, P. V.; Xue, D.; Yuan, R. Active learning in materials science with emphasis on adaptive sampling using uncertainties for targeted design. NPJ. Comput. Mater. 2019, 5, 21.
152. Ueno, T.; Rhone, T. D.; Hou, Z.; Mizoguchi, T.; Tsuda, K. COMBO: An efficient Bayesian optimization library for materials science. Mater. Discov. 2016, 4, 18-21.
153. Takeda, H.; Murakami, K.; Yamaguchi, Y.; et al. Experimental data-driven efficient exploration of the composition and process conditions of Li-rich NASICON-type solid electrolytes. Next. Mater. 2025, 8, 100574.
154. Fukuda, H.; Kusakawa, S.; Nakano, K.; et al. Bayesian optimisation with transfer learning for NASICON-type solid electrolytes for all-solid-state Li-metal batteries. RSC. Adv. 2022, 12, 30696-703.
155. Takeda, H.; Fukuda, H.; Nakano, K.; et al. Process optimisation for NASICON-type solid electrolyte synthesis using a combination of experiments and Bayesian optimisation. Mater. Adv. 2022, 3, 8141-8.
156. Matsuda, S.; Lambard, G.; Sodeyama, K. Data-driven automated robotic experiments accelerate discovery of multi-component electrolyte for rechargeable Li-O2 batteries. Cell. Rep. Phys. Sci. 2022, 3, 100832.
157. Whitacre, J. F.; Mitchell, J.; Dave, A.; Wu, W.; Burke, S.; Viswanathan, V. An autonomous electrochemical test stand for machine learning informed electrolyte optimization. J. Electrochem. Soc. 2019, 166, A4181-7.
158. Dave, A.; Mitchell, J.; Kandasamy, K.; et al. Autonomous discovery of battery electrolytes with robotic experimentation and machine learning. Cell. Rep. Phys. Sci. 2020, 1, 100264.
159. Stolberg, M. A.; Lopez, J.; Cawthern, S. D.; et al. A data-driven platform for automated characterization of polymer electrolytes. Matter 2025, 8, 102129.
160. Boiko, D. A.; MacKnight, R.; Kline, B.; Gomes, G. Autonomous chemical research with large language models. Nature 2023, 624, 570-8.
161. Kamikawa, Y.; Amezawa, K.; Terada, K. Chemo-electro-mechanical phase-field simulation of interfacial nanodefects and nanovoids in solid-state batteries. Commun. Mater. 2024, 5, 180.
162. You, Y.; Zhang, D.; Wu, Z.; et al. Grain boundary amorphization as a strategy to mitigate lithium dendrite growth in solid-state batteries. Nat. Commun. 2025, 16, 4630.
163. Huo, H.; Bai, Y.; Benz, S. L.; et al. Decoupling the effects of interface chemical degradation and mechanical cracking in solid-state batteries with silicon electrode. Adv. Mater. 2025, 37, e2415006.
164. Rajagopal, D.; Koeppe, A.; Esmaeilpour, M.; et al. Data-driven virtual material analysis and synthesis for solid electrolyte interphases. Adv. Energy. Mater. 2023, 13, 2301985.
165. De Baas, A.; Nostro, P. D.; Friis, J.; et al. Review and alignment of domain-level ontologies for materials science. IEEE. Access. 2023, 11, 120372-401.
166. Dreger, M.; Eslamibidgoli, M. J.; Eikerling, M. H.; Malek, K. Synergizing ontologies and graph databases for highly flexible materials-to-device workflow representations. J. Mater. Inf. 2023, 3, 2.
167. Stier, S. P.; Xu, X.; Gold, L.; Möckel, M. Ontology-based battery production dataspace and its interweaving with artificial intelligence-empowered data analytics. Energy. Tech. 2024, 12, 2301305.
168. Mutz, M.; Perovic, M.; Gümbel, P.; et al. Toward a Li‐ion battery ontology covering production and material structure. Energy. Tech. 2022, 11, 2200681.
169. Clark, S.; Battaglia, C.; Castelli, I. E.; et al. Semantic resources for managing knowledge in battery research. ChemSusChem 2025, 18, e202500458.
170. Clark, S.; Bleken, F. L.; Stier, S.; et al. Toward a unified description of battery data. Adv. Energy. Mater. 2021, 12, 2102702.
171. Ward, L.; Babinec, S.; Dufek, E. J.; et al. Principles of the battery data genome. Joule 2022, 6, 2253-71.
172. Ghedini, E.; Goldbeck, G.; Friis, J.; et al. European Materials & Modelling Ontology. https://github.com/emmo-repo/EMMO.
173. Del Nostro, P.; Goldbeck, G.; Toti, D. CHAMEO: An ontology for the harmonisation of materials characterisation methodologies. AO. 2022, 17, 401-21.
174. Del Nostro, P.; Goldbeck, G.; Kienberger, F.; et al. Battery testing ontology: An EMMO-based semantic framework for representing knowledge in battery testing and battery quality control. Computers. in. Industry. 2025, 164, 104203.
175. Venugopal, V.; Olivetti, E. MatKG: An autonomously generated knowledge graph in Material Science. Sci. Data. 2024, 11, 217.
176. Bai, J.; Mosbach, S.; Taylor, C. J.; et al. A dynamic knowledge graph approach to distributed self-driving laboratories. Nat. Commun. 2024, 15, 462.
177. Rihm, S. D.; Tan, Y. R.; Ang, W.; et al. The Digital Lab Facility Manager: Automating operations of research laboratories through “The World Avatar”. Nexus 2024, 1, 100031.
178. Bai, J.; Cao, L.; Mosbach, S.; Akroyd, J.; Lapkin, A. A.; Kraft, M. From platform to knowledge graph: evolution of laboratory automation. JACS. Au. 2022, 2, 292-309.
179. Wang, S.; Liu, J.; Song, X.; et al. Artificial intelligence empowers solid-state batteries for material screening and performance evaluation. Nanomicro. Lett. 2025, 17, 287.
180. Wang, K.; Gupta, V.; Lee, C. S.; et al. XElemNet: towards explainable AI for deep neural networks in materials science. Sci. Rep. 2024, 14, 25178.
181. Tao, K.; Li, J.; He, W.; et al. CGformer: Transformer-enhanced crystal graph network with global attention for material property prediction. Matter 2025, 8, 102380.
182. Wang, Y.; Shi, J.; Gao, H.; Wang, M. S.; Lin, C. Study of void evolution in lithium solid-state batteries: integrating high-throughput phase-field modeling, experimental validation, and machine learning. Adv. Energy. Mater. 2025, 15, 2501616.
183. Liu, B.; Yang, J.; Yang, H.; et al. Rationalizing the interphase stability of Li|doped-Li7La3Zr2O12 via automated reaction screening and machine learning. J. Mater. Chem. A. 2019, 7, 19961-9.
184. Hwang, H.; Jeong, H.; Cho, J. W.; et al. Machine learning-assisted microstructural quantification of multiphase cathode composites in all-solid-state batteries: correlation with battery performance. Small 2025, 21, e2410016.
185. Ding, K.; Yu, J.; Huang, J.; Yang, Y.; Zhang, Q.; Chen, H. SciToolAgent: a knowledge-graph-driven scientific agent for multitool integration. Nat. Comput. Sci. 2025, 5, 962-72.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.