


Atherosclerosis in women: immune regulation under sex differences
 0
0 
Abstract
Atherosclerosis (AS) is a chronic inflammatory disease initiated by abnormal subendothelial lipid deposition in blood arteries. Although endothelial dysfunction, lipid homeostasis imbalance, and clonal proliferation of vascular smooth muscle cells are central to classical theories, recent studies have shown that immune cells such as neutrophils, T lymphocytes, and macrophages dynamically infiltrate the plaque microenvironment. This evidence suggests that the immune regulatory network plays a significant role in the pathological process of AS. It should be noted that female patients exhibit distinct clinical phenotypes and pathological characteristics during AS. This sex difference is closely related to changes in sex hormone levels, such as estrogen, as well as to differences in inflammatory responses and metabolic regulation. Studies aiming to control the progression of AS by targeting immune mechanisms unique to women remain in the exploratory stage, and their potential mechanisms of action and regulation have not yet been well clarified. This study will systematically elucidate immune activity and provide a theoretical basis for effective intervention strategies for cardiovascular disease in women, focusing on immunological remodeling and the relationship between sex and immunity in AS.
Keywords
INTRODUCTION
Atherosclerotic cardiovascular disease (ASCVD), the leading cause of death and disability worldwide, remains critically important to prevent and control. According to World Health Organization data, cardiovascular diseases claimed approximately 18 million lives globally in 2019, accounting for 32% of all deaths. Epidemiological evidence indicates that the incidence of ASCVD continues to rise, although modern clinical interventions have significantly reduced the adverse effects of traditional risk factors such as smoking, diabetes, hyperlipidemia, and hypertension[1,2].
Men have greater incidence and mortality rates of ASCVD than women in the general population. While women often have a marked increase in ASCVD incidence 7-10 years after menopause, males typically develop the syndrome earlier (40-60 years)[3]. This phenomenon is intimately linked to
This review methodically clarifies the pathophysiological features of atherosclerosis from the viewpoints of immune regulatory systems and gender differences, addressing the shortcomings of existing studies. It explores the molecular and cellular signaling pathways that underlie the relationship between sex hormones and the immune system, exposing their possible roles in immune cell activation and polarization as well as vascular inflammation. Establishing a theoretical framework for precision prevention and treatment approaches aimed at cardiovascular illnesses unique to women is the goal.
THE ROLE OF THE IMMUNE SYSTEM IN ATHEROSCLEROSIS
Rudolf Virchow identified the presence of immune cells in plaques and provided the first immunological explanation of atherosclerosis in the middle of the 19th century. After Jonasson et al. separated T cells from human plaques using immunofluorescence, “atherosclerosis immunology” became a recognized field of research. This revealed the presence of adaptive immune responses in atherosclerosis for the first time[4]. This idea shows that atherosclerosis involves both the innate and adaptive immune systems and is an active inflammatory process that is brought on by endothelial injury rather than a passive lipid buildup.
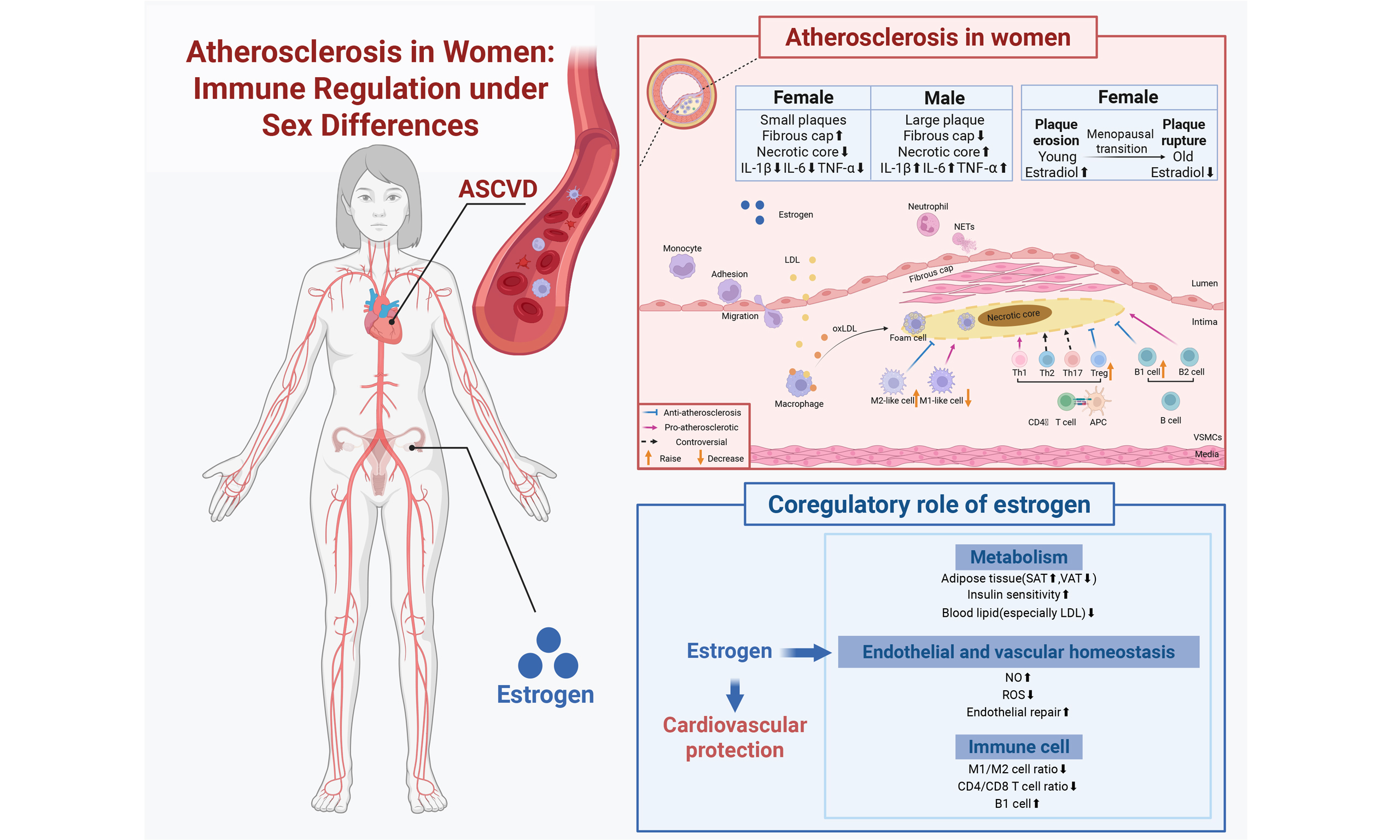
In short, circulating lipoproteins such as low-density lipoprotein (LDL) can get into and stay in the area just below the damaged endothelium caused by atherosclerosis. These lipoproteins, including their oxidized versions such as oxidized LDL (oxLDL), encourage vascular smooth muscle cells and endothelial cells to make substances such as monocyte chemoattractant protein 1 (MCP-1), vascular cell adhesion molecule 1 (VCAM1) and intercellular adhesion molecule 1 (ICAM1), which draw monocytes and other inflammatory cells to the artery wall [Figure 1].
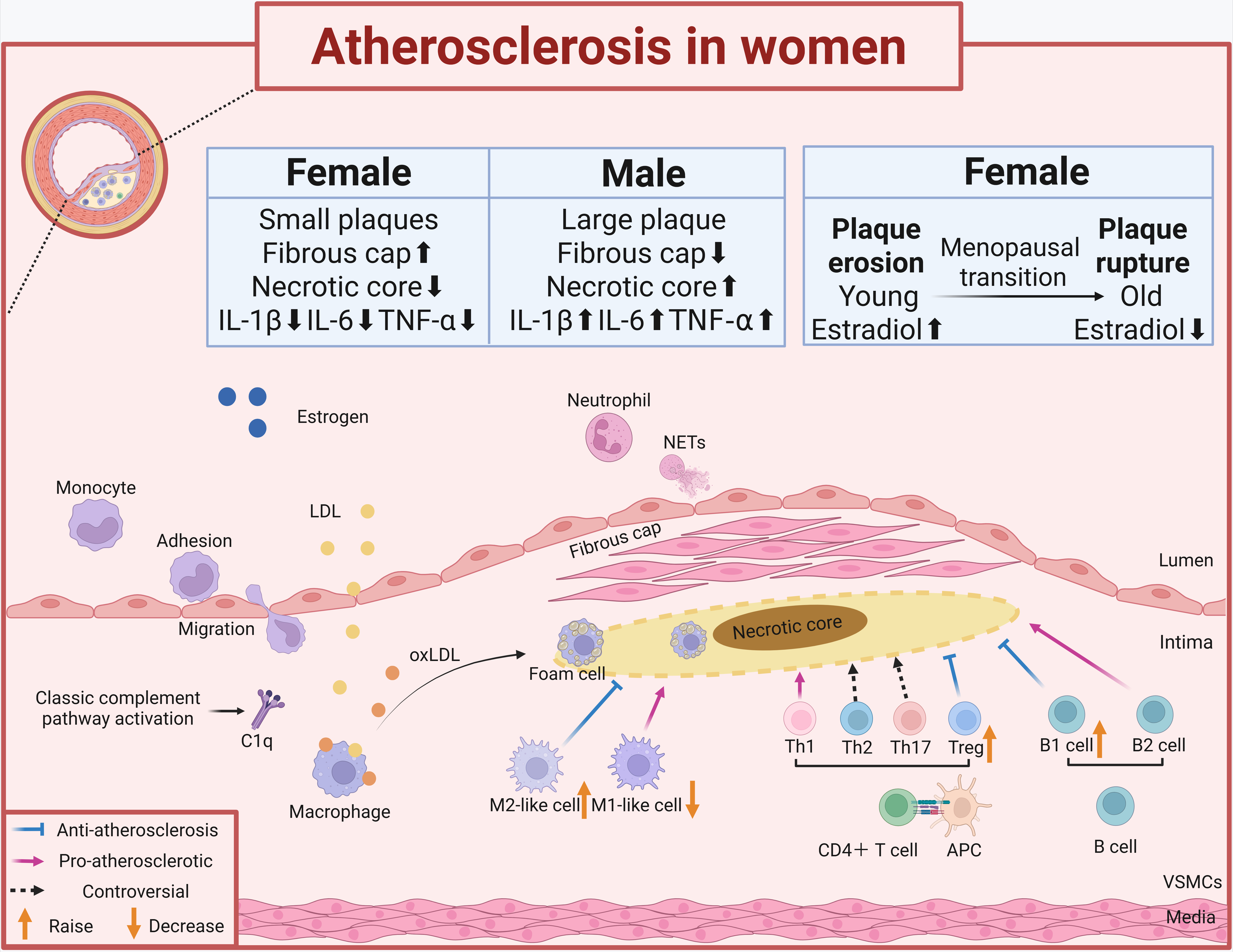
Figure 1. The role of the immune system in atherosclerosis in women. We demonstrate the sex-specific immune mechanisms of atherosclerosis in women, highlighting the association of immune regulation with estrogen action. The boxed content compares plaque differences between men and women, showing the sex-specific features of inflammatory cell (e.g., macrophage, T-cell, and B-cell) infiltration in women’s plaques. IL-1β: Interleukin-1β; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α; LDL: low-density lipoprotein; NETs: neutrophil extracellular trapping networks; oxLDL: oxidized LDL; Treg: regulatory T cells; VSMCs: vascular smooth muscle cells. The figure was created with https://app.biorender.com/illustrations/684df97250730a2a26c9c59c.
Innate immunity
Early atherogenesis triggers the activation of innate immunity, the body’s earliest defense against infections. In reaction to chemokines, the number of circulating monocytes rises under the influence of hypercholesterolemia, and they are drawn to the arterial wall[5]. Atherosclerosis is primarily caused by classical monocytes, which constitute the majority of monocytes recruited to atherosclerotic plaques. These monocytes cause more inflammation than non-classical monocytes and are regulated by the chemokine receptor C-C chemokine receptor 2 (CCR2) and its main partner C-C chemokine ligand 2 (CCL2)[6].
Macrophage
As essential immune cells in atherosclerosis, macrophages are crucial for the development, maintenance, and reduction of inflammation in Atherosclerosis plaques. Three types of macrophages can be found at atherosclerotic plaques: (1) circulating monocytes, (2) vascular smooth muscle cells that have undergone transdifferentiation, and (3) artery-resident macrophages. The principal contributors to atherosclerosis are inflammatory macrophages, which mostly come from circulating monocytes[7]. Early plaque lesions are characterized by lipid-rich foam cells that are progressively formed by the differentiation of monocytes into macrophages, which phagocytose and eliminate lipoproteins. Too many foam cells eventually lead to cell death, which causes inflammation and forms a dead tissue area, speeding up the process of atherosclerosis.
Macrophages can have both pro- and anti-inflammatory effects, demonstrating their significant functional variability. Based on in vitro experiments, early studies merely divided macrophages into two subtypes: type M1 macrophages, which are induced by lipopolysaccharide (LPS) and interferon gamma (IFN-γ), secrete pro-inflammatory factors such as interleukin-1β (IL-1β), interleukin (IL)-6 and tumor necrosis factor (TNF)-α, which have strong pro-inflammatory and bactericidal effects; and type M2 macrophages, which are induced by IL-4, IL-10 and IL-13, secrete anti-inflammatory factors such as IL-10 and TGF-β, which support tissue remodeling and repair. The actual macrophage kinds found in the human body are not reflected in this classification, which is nonetheless considered traditional. There may be a more varied distribution of macrophage subpopulations within atherosclerotic plaques due to the complexity of the plaque microenvironment and the high degree of macrophage flexibility. Through the use of cutting-edge technologies such as single-cell RNA sequencing (scRNA-seq), several macrophage subpopulations have been identified in atherosclerotic plaque lesions. These subpopulations exhibit notable variations in their gene expression levels and functions[8,9]. Typically, pro-inflammatory macrophages have distinct gene expression profiles, such as increased transcript levels of pro-inflammatory cytokines (IL-6, TNF-α and
Neutrophil
The most prevalent leukocytes in the bloodstream, neutrophils were once assumed to be merely defensive leukocytes. But now, several “subpopulations” of neutrophils are thought to exist, each with a unique role in the bloodstream and other tissues[11,12]. According to epidemiologic studies, the risk of ASCVD is strongly predicted by an elevated neutrophil lymphocyte ratio (NLR)[13]. The peripheral neutrophilia caused by hypercholesterolemia is regulated by increasing bone marrow mobilization, lowering clearance, and encouraging granulopoiesis[14]. It also plays a significant role in the development of early atherosclerotic lesions. Damage-associated molecular patterns (DAMPs) cause N2-type neutrophils (Ly6G+ CD206+) to reprogram into pro-inflammatory N1-type neutrophils (Ly6G+ CD206-) during myocardial infarction through Toll-like receptor 4 (TLR4)[15]. Neutrophils not only cause atherosclerosis by infiltrating the arterial wall and releasing inflammatory mediators, but additional research has indicated that they also worsen thrombosis and inflammation in atherosclerosis by forming neutrophil extracellular trapping networks (NETs) and microvesicles[16-19].
Dendritic cell
Dendritic cells (DCs), important antigen-presenting cells (APCs), link innate and adaptive immunity. Mature DCs travel to the lymph nodes from their bone marrow origins, where they show improved responses to particular chemokines and a greater capacity to stimulate T cell proliferation[20]. Tissue-resident DCs are immature at homeostasis, and triggers such as cytokines or DAMPs can activate immature DCs to mature cells[21]. AS progresses as activated DCs present antigens to T cells to start an adaptive immune response[22].
Complement system
An essential part of innate immunity, the complement system acts as a vital link between innate and adaptive immunity. Comprising a number of plasma proteins, it is triggered via the lectin, alternative, and classical pathways. This ultimately results in the release of inflammatory mediators, the cleavage of complement components, and the creation of the membrane assault complex. Furthermore, as a fourth channel for complement activation, the coagulation system may contribute to the development of atherosclerosis. Atherosclerosis is influenced by a number of complement components, each of which plays a dual role in either promoting or inhibiting the process. The proteolytic cleavage of the core complement component C3, which acts as the site of convergence for additional effector actions across pathways, is essential to all three complement activation pathways. Complement components at different activation levels, such as C1q, C5a, and C5b-9 complement complexes, were later discovered after early research showed markedly higher C3 protein concentrations in atherosclerotic plaque tissue relative to normal artery intima[23].
Complement C1q, as an initiator of the classical activation pathway, plays a dual role in AS[24]. By binding pattern recognition molecules such as modified lipoproteins, cholesterol crystals, or C-reactive protein, as well as by identifying autoantibodies against oxLDL, C1q triggers the classical complement cascade. It produces active products such as C3a, C5a, and the C5b-9 complement complex after the complement system is activated by the classical pathway. These products may accelerate the development of atherosclerosis. The Nod-like receptor protein 3 (NLRP3) inflammasome is activated by the C5b-9 complement complex, and it then acts on macrophages and endothelial cells to worsen vascular inflammation[25]. However, low-density lipoprotein receptor knockout (LDLR-/-) mice with C1q gene abnormalities show more apoptotic cells and larger aortic root lesions than LDLR-/- mice[26]. C1q exerts anti-atherosclerotic effects by inhibiting the secretion of pro-inflammatory cytokines IL-6 and IL-1β[27]. Additionally, C1q inhibits the development of atherosclerosis by reducing the production of foam cells and increasing cholesterol efflux from macrophages[28]. Strong anaphylatoxins C3a and C5a cause mast cell degranulation, which releases cytokines and histamine, increases vascular permeability, and chemotaxes T cells and macrophages into the plaque[29].
In conclusion, by attracting immune cells, triggering inflammatory reactions, improving antigen presentation, and regulating immune cell responses, the complement system contributes to the onset and advancement of atherosclerosis. Although some components, such as C1q, have bidirectional regulatory effects, research suggests that the complement system mostly supports AS. Thus, pinpointing the precise processes via which the complement system causes atherosclerosis can help determine potential treatment options for AS.
Adaptive immunization
Autoimmunity, exemplified by the loss of immune tolerance to self-antigens (e.g., the production of autoantibodies against lipoproteins), is a hallmark of the chronic inflammatory process of atherosclerosis[30]. This phenomenon is closely associated with the activation of adaptive immune cells, specifically T and B cells.
T cell
T cells, which are primarily classified as cytotoxic T cells (CD8+ T cells) and helper T cells (CD4+ T cells), are a crucial part of the adaptive immune system. CD4+ T cells play a central role in mediating the atherosclerotic inflammatory response by assisting in the maturation of B cells and the production of antibodies, while at the same time rendering CD8+ T cells cytotoxic. APCs contain MHC class II molecules, which CD4+ T lymphocytes recognize to become activated. It is important to further differentiate into distinct subpopulations, such as pro-inflammatory Th1/Th17 and anti-inflammatory Th2/regulatory T cells (Treg). CD4+ T cells can develop into Treg and a range of functionally diverse helper T cells [Th1, Th2, Th9, Th17, Th22, follicular helper T cells (Tfh), and CD28null T cells][31]. It is believed that a major contributing factor to the development of atherosclerosis is the imbalance between different CD4+ T cell subsets[32].
Th1 and Th2 cells constituted the majority of the CD4+ T cell population invaded by atherosclerotic plaques, with the TH1 cell subset predominating[33]. Th1 cells express the T-box transcription factor TBX21 (T-bet) and chemokine receptors (CXCR3 and CCR5). Th1 cells secrete the pro-inflammatory cytokine IFN-γ, which promotes the development of atherosclerosis. IFN-γ deficiency significantly attenuates the extent of atherosclerotic lesions in mice, independently of serum lipoprotein profile[34]. Th17 cells release IL-17, causing more inflammation[35], but it also helps produce collagen in blood vessel muscle cells and makes plaques more stable[36]. Th2, which is related to IL-4, is generally considered to be atheroprotective, although its protective role is controversial due to its role in mediating anaphylactic responses.
Treg cells are an important subpopulation of anti-atherosclerotic cells that exert their protective effects by inhibiting T cell proliferation and secreting anti-inflammatory cytokines (TGF-β and IL-10)[37]. Treg cells inhibit overreactive immune responses in atherosclerosis, limiting persistent inflammation and autoimmunity. Increasing the quantity or functionality of Treg cells can partially prevent and treat atherosclerosis[38]. As a new immunomodulatory approach to cardiovascular disease, tolerant vaccination works to prevent atherosclerosis by reestablishing the balance of immunological tolerance and selectively stimulating the growth of Treg cells that target antigens linked to atherosclerosis. However, a related concern is that Treg cells might shift from initially promoting a protective immune response to instead supporting a harmful one. This is because forkhead box P3 (Foxp3)-expressing Treg cells are functionally flexible, and when they do not produce Foxp3 regularly, they become unstable, progressively losing their capacity to suppress the immune system and becoming ex-Treg cells[39].
There are several lines of evidence supporting the pro-atherosclerotic effects of Tfh, which mainly encourage the establishment of germinal centers and the switching of B cell antibody classes[40,41]. More thorough mechanistic research is required to examine the pro-atherosclerotic or protective roles of T cell subsets, including Th9, Th22 and γδT, as their precise role in atherosclerosis remains debated.
B cell
Another significant adaptive immune cell type, the B lymphocyte, plays dual roles in atherosclerosis. By modifying T cell responses and producing cytokines and antibodies, B cells influence atherogenesis[42]. The two primary B-cell lineages are B1 cells, which can be further classified into B1a and B1b subpopulations, and B2 cells, which can differentiate into follicular (FO) and marginal zone (MZ) cells[43]. These subsets play distinct roles in B cell-mediated atherosclerosis.
Early research established the existence of specific immunoglobulins that target various oxLDL epitopes in atherosclerotic plaques. These epitopes, known as oxidation-specific epitopes (OSEs), are created when LDL undergoes oxidative alteration. The atherosclerotic process produces both natural and adaptive antibodies that primarily target these epitopes[44]. While B2 cells contribute to atherosclerosis by producing Immunoglobulin G (IgG), secreting the pro-inflammatory cytokine TNF-α, and activating CD4+ T cells to promote atherosclerosis[45], B1 cells create Immunoglobulin M (IgM) that is directed against the OSEs on LDL, protecting against AS[46]. Other autoantibodies that contribute to the development of atherosclerosis include anti-cyclic citrullinated peptide antibodies, anti-apolipoprotein A-I antibodies, and anti-heat shock protein 60 (HSP60) antibodies.
So, treatments that remove B cells, including anti-CD20 antibodies and anti-B cell-activating factor (BAFF) antibodies, which focus on different types of B cells, could help with atherosclerosis in humans.
IMMUNOLOGIC SPECIFICITY OF ATHEROSCLEROSIS IN WOMEN
Characteristics of atherosclerosis in women
Trend of incidence
Women typically develop ASCVD at a late age, frequently following menopause, which is evidently linked to a considerable drop in estrogen levels. During menopause, a woman’s ovarian function steadily diminishes until it stops entirely. This drop, coupled with decreased estrogen levels, affects the body’s immune system and metabolism[47-49]. Compared to premenopausal women, postmenopausal women have a roughly fourfold higher prevalence of coronary heart disease[50] and a significantly higher risk of metabolic illnesses, including obesity and diabetes[51]. In aged LDLR-/- mice (22 months old), females exhibited a higher M1/M2 macrophage ratio and a more pronounced pro-inflammatory macrophage phenotype, characterized by elevated expression of NLRP3, C-X-C motif chemokine ligand 2 (CXCL2), and matrix metalloproteinase 9 (MMP9) within atherosclerotic plaques, compared to aged males[52].
Nonetheless, research indicates that the risk of ASCVD in women of reproductive age is often underrecognized. This is partly because previous studies have primarily focused on men and postmenopausal women, who have a higher incidence of ASCVD, and partly because women of reproductive age may present with atypical angina symptoms, leading to delayed diagnosis. According to a study that looked at the cardiac autopsy of 87 women between the ages of 15 and 50, women’s markedly elevated risk of ASCVD can start as early as age 30. The left anterior descending artery is most commonly impacted, though it usually affects many coronary arteries[53]. This implies that although estrogen has heart-healthy preventive effects on women of reproductive age, younger women’s cardiovascular health is still influenced by other factors.
Risk factors
While smoking, high blood pressure, dyslipidemia, diabetes, physical inactivity, and obesity are among the many classic cardiovascular risk factors that men and women have in common, their effects differ by sex. Although men smoke significantly more than women worldwide, smoking poses a greater risk to women’s cardiovascular health. The mortality rate from coronary heart disease is 25% higher in female smokers than in male smokers[54,55].
Although men are more likely than women to have hypertension, women’s blood pressure rises more quickly after age 30. The prevalence of hypertension increases after menopause and may overtake that of males in later life[56]. This is related to estrogen’s regulation of vasodilation mediated by the
Women’s blood lipid levels, particularly LDL cholesterol, are noticeably higher during or after menopause, which raises their risk of atherosclerosis[58-60]. Compared to men of the same age, premenopausal women had higher levels of high-density lipoprotein cholesterol (HDL-C). Low-density lipoprotein cholesterol
Men are more likely to store fat in visceral locations [visceral adipose tissue (VAT)] than women, who are more likely to store fat in subcutaneous areas such as the thighs and buttocks [subcutaneous adipose tissue (SAT)][62]. This discrepancy is directly linked to how estradiol controls adipocyte function and skeletal muscle lipid oxidation[63]. When transgender people received large dosages of estrogen, their legs and feminized areas had more subcutaneous adipose tissue[64]. There is a tight relationship between visceral fat accumulation, insulin resistance, and atherosclerosis. Because women’s estrogen levels decrease after menopause, visceral fat accumulation increases and the incidence of diabetes rises, which can hasten the production of atherosclerotic plaques[65]. Additionally, studies show that women with type 2 diabetes have a far higher adjusted hazard ratio (HR) for fatal coronary artery disease than males do[66].
Furthermore, risk factors unique to women should be considered, including polycystic ovary syndrome (PCOS), gestational diabetes, autoimmune diseases, and hypertensive disorders of pregnancy (such as preeclampsia). Younger women are more susceptible to ASCVD because of these risk factors, which mainly affect women of reproductive age. These gender-specific factors can affect women’s cardiovascular health earlier and increase the risk of ASCVD after the dramatic drop in estrogen levels during menopause, in contrast to traditional risk factors that benefit from estrogen protection. For instance, women with autoimmune illnesses are around 40% more likely than healthy people to experience a cardiovascular incident[67]. According to another study, women with systemic lupus erythematosus (SLE) between the ages of 35 and 44 had a 50-fold increased risk of myocardial infarction compared to women without SLE of the same age[68]. Women with autoimmune illnesses are far more negatively affected by the postmenopausal drop in estrogen, and research indicates that women with SLE are three to five times more likely than the general population to experience cardiovascular problems following menopause[69].
Plaque characteristics
Plaque morphology differs significantly between men and women. Female patients show less macrophage infiltration and smaller necrotic cores within plaques than male patients[70]. Intraplaque bleeding and
Studies on pathology have shown that plaque erosion - characterized by endothelial cell detachment, enrichment of smooth muscle cells and proteoglycans, and reduced inflammatory cell infiltration - is the primary pathological basis in young female patients with acute myocardial infarction, accounting for more than 80% of cases in women under 50[71]. In contrast to plaque rupture, eroded plaques are characterized by an intact fibrous cap. Thrombosis in this context originates from endothelial denudation, which leads to the exposure of underlying collagen. The mechanistic pathway involves Toll-like receptor 2 (TLR2)-mediated release of neutrophil extracellular traps (NETs), and NET-induced endothelial apoptosis. These processes collectively promote the formation of a predominantly platelet-rich (white) thrombus[72]. In postmenopausal women, the fibrous cap thins, the necrotic core of plaques gradually enlarges, and the likelihood of plaque rupture increases due to variables such as age and decreased estrogen levels.
Composition of immune cells
The ability to resolve sex differences in immune cells at ultra-high resolution has been made possible by recent high-throughput technologies, especially scRNA-seq. Huang et al. revealed that sex influences the composition and function of immune cells in the peripheral blood of healthy adults: males exhibit a significantly higher proportion of natural killer (NK) cells compared to females, while females demonstrate a higher proportion of plasma cells. Concurrently, females display stronger adaptive immune response characteristics, manifested by enhanced B-cell activation signaling pathways [activation of BAFF and A Proliferation-Inducing Ligand (APRIL) signaling pathways][73].
Sukhavasi et al. analyzed human carotid plaques and identified significant sex differences in smooth muscle cell, endothelial cell, and macrophage subtypes. The study performed deep single-cell sequencing on carotid plaque cells from 7,690 male and female patients. Although there were no discernible changes between the main cell types in male and female atherosclerotic plaques, notable variances were found in their cellular phenotypes. Significant sex bias was seen in three endothelial cell subpopulations, six smooth muscle cell subpopulations, and six macrophage subpopulations. The role of smooth muscle contraction was regulated by the primary contractile phenotype of male smooth muscle cells. Angiogenesis and T cell-mediated cytotoxicity were linked to endothelial cell activity, whereas tissue remodeling was linked to macrophages. In female patients, macrophages showed a lipid-related inflammatory phenotype with high
There is currently a dearth of research on identifying macrophage subpopulations in the atherosclerotic process in women. On the one hand, the majority of studies only identify subpopulations based on gene expression profiles, and there are no systematic protein marker analyses or functional validation. On the other hand, there are not many studies on the heterogeneity of macrophage subpopulation distributions and functions in the pathologic microenvironments unique to women. In order to clarify the molecular reasons behind the gender differences, future research should concentrate on analyzing the subpopulation composition and functional traits of macrophages in female atherosclerotic plaques.
In terms of adaptive immune cell composition, a study using single-cell sequencing technologies such as scRNA-Seq and Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-Seq) found that male and female patients with diabetes and coronary artery disease had significantly different gene expression patterns in their CD4+ T cells. CCR2+ effector memory (Em), MMP9+, and programmed
Gender dimorphism is also seen in the cytokine profile of peripheral blood. The levels of these
Immunoregulatory effects of sex hormones
Males and females have distinct innate and adaptive immune systems; some differences persist throughout life, while others emerge after puberty and before reproductive decline, highlighting the critical role of sex hormones. These immunological sex differences, manifested as variations in vascular inflammation and immune activation, not only influence the prevalence of autoimmune diseases and cancers but also strongly regulate the development of atherosclerosis. Estrogens, androgens, and progestogens can bind to extracellular and intracellular receptors through ligand-dependent, ligand-independent, genomic, or
Estrogen
Estrone (E1), estradiol (E2), and estriol (E3) are the three main types of estrogens. Granulosa cells in the ovaries create estradiol, the main form of estrogen in circulation in women of reproductive age. Estradiol levels drop precipitously in postmenopausal women, and estrone takes over as the main type of estrogen. Pregnancy causes a sharp increase in estriol levels, which indicate placental activity. E2 levels rise during pregnancy, fall sharply after menopause, and fluctuate during the female menstrual cycle. The density, distribution, and kind of estrogen receptors in immune cells all affect how estrogen affects immune function in addition to its concentration. As of right now, there are three known estrogen receptors: the G
Numerous lines of evidence indicate estrogen’s preventive function in preserving vascular and endothelial homeostasis in atherosclerosis. Estradiol activates nitric oxide synthase in endothelial cells, which leads to the release of nitric oxide and encourages vasodilation[78]. After estrogen therapy, vascular smooth muscle cells release substances that stop epithelial fibroblast movement, which helps in the formation of neointima[79]. ERα expression and vascular endothelial autophagy are lower in postmenopausal women than in premenopausal women. By increasing the expression of ERα and triggering autophagy, estrogen protects against atherosclerosis by lowering NLRP3 inflammasome-IL1β-mediated inflammation and vascular endothelial cell death[80]. In premenopausal women, estradiol prevents vascular inflammation and atherosclerosis, lessens oxidative stress, and lowers the generation of ROS[81-83]. Estradiol-treated ovariectomized rats exhibited improved vascular remodeling due to a significant decrease in the expression of the adhesion molecules ICAM1, VCAM1, and P-selectin, as well as a decrease in the levels of the
Through various pathways, such as the control of lipid metabolism, inflammatory reactions, oxidative stress, and cell survival, estrogen affects macrophage activity [Figure 2]. Macrophages play a major role in the formation of foam cells, which is a crucial early event in atherosclerosis. The lectin-like oxidized
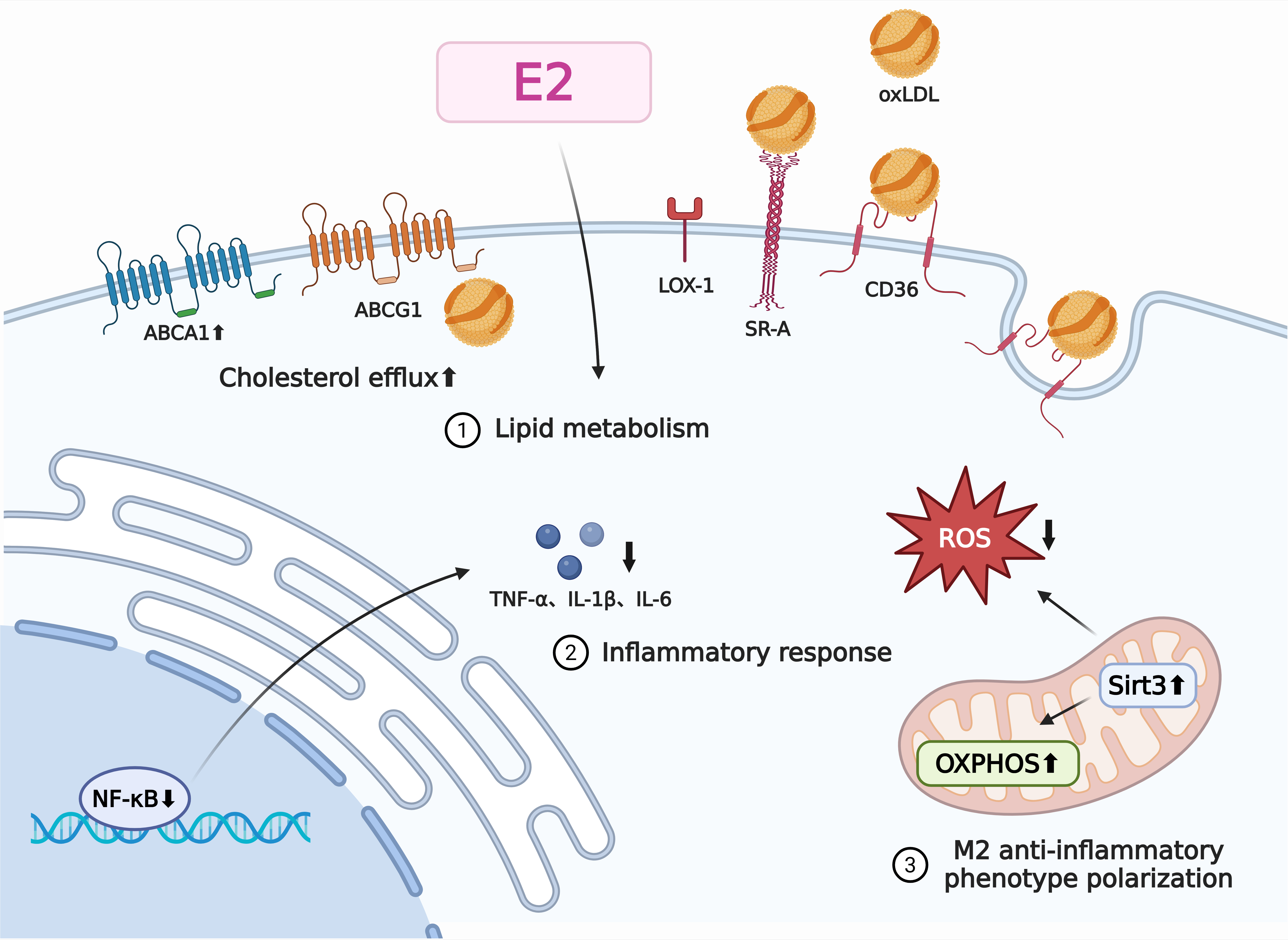
Figure 2. The regulatory mechanism of estrogen on macrophages in atherosclerosis. We have elucidated the mechanisms by which estrogen regulates macrophage function in atherosclerosis. Estrogen influences macrophage function through its effects on lipid metabolism, inflammatory responses, and cellular polarization. The specific mechanism of action is as follows: Estrogen promotes cholesterol efflux from macrophages, thereby reducing foam cell formation and delaying the progression of atherosclerosis; estrogen inhibits NF-κB activation, reducing production of pro-inflammatory cytokines TNF-α, IL-1, and IL-6; and estrogen induces metabolic reprogramming in macrophages (enhancing mitochondrial respiration while decreasing reactive oxygen species production), thereby promoting their conversion to anti-inflammatory M2 macrophages. IL-1β: Interleukin-1β; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α; oxLDL: oxidized low-density lipoprotein; ABCA1: ATP-binding cassette transporter A1; ABCG1: ATP-binding cassette transporter G1; SR-A: scavenger receptor A; LOX-1: lectin-like oxidized low-density lipoprotein receptor 1; ROS: reactive oxygen species; OXPHOS: oxidative phosphorylation; Sirt3: sirtuin 3; NF-κB: nuclear factor kappa B. The figure was created with https://app.biorender.com/illustrations/68b8e4b10db6ea9d6bb9adb4.
One important area of study is how estrogen affects macrophage polarization. Studies have shown that estradiol can reduce the pro-inflammatory phenotype of M1 macrophages, inhibit the production of reactive oxygen species (ROS) in the mitochondria, and encourage their transformation into
Estrogen specifically influences the immunological activity of macrophages by inhibiting the production of pro-inflammatory cytokines. By preventing nuclear factor kappa B (NF-κB) activation in macrophages, estradiol can reduce the amount of TNF-α generated by LPS[90]. Estrogen can prevent the production of
As the body’s primary system for generating free radicals, neutrophils are essential for the development of atherosclerotic plaques and other free radical-mediated disorders. By controlling neutrophil activation and superoxide generation, estrogen indirectly reduces inflammation and promotes antioxidants. Marczell et al. showed that 17-β-estradiol inhibits the formation of neutrophil superoxide, a crucial regulator of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, by targeting the Rac1 protein[96]. The anti-inflammatory mechanism of estradiol has been further clarified by recent research: E2 dephosphorylates the extracellular signal-regulated kinase (ERK) pathway and upregulates the protein levels of mitogen-activated protein kinase phosphatase 2 (MKP-2) to prevent N-formyl-Met-Leu-Phe (fMLP)-induced migration and superoxide production in human neutrophils and HL-60 differentiated neutrophil-like cells (dHL-60)[97].
Previous research suggests that estrogen may control the creation of NETs, influencing the process in both directions and, depending on the particular microenvironment, either promoting or inhibiting it. According to studies, E2 administration causes NETosis (Neutrophil Extracellular Traps formation) through
T cells also express ERs in large quantities, and several studies show that estrogen largely controls T cell differentiation and proliferation to affect the development of atherosclerosis. In mouse models of ovariectomized (OVX) mice, CD4+ T cells proliferate when estrogen is deficient, whereas CD4+ T cells undergo apoptosis when exposed to estrogen[100]. Estrogen can affect T cells through GPER. CD4+ T cells express more IL-10 when GPER is activated[101]. Additionally, research has demonstrated that GPER activation can increase Treg cells by inducing the expression of the transcription factor FOXP3 following CD4+ T cell polarization into TH17 cells in vitro[102]. According to research, estrogen has a dose-dependent influence on CD4+ T cell polarization. While high amounts of estrogen (during pregnancy or pharmacological settings) inhibit Th1-driven cellular immune responses, low levels of estrogen (at physiological levels) enhance IFN-γ production[103,104]. The main mechanism by which E2 exposure promotes Th1-to-Th2 differentiation is ERα activation [Figure 3].
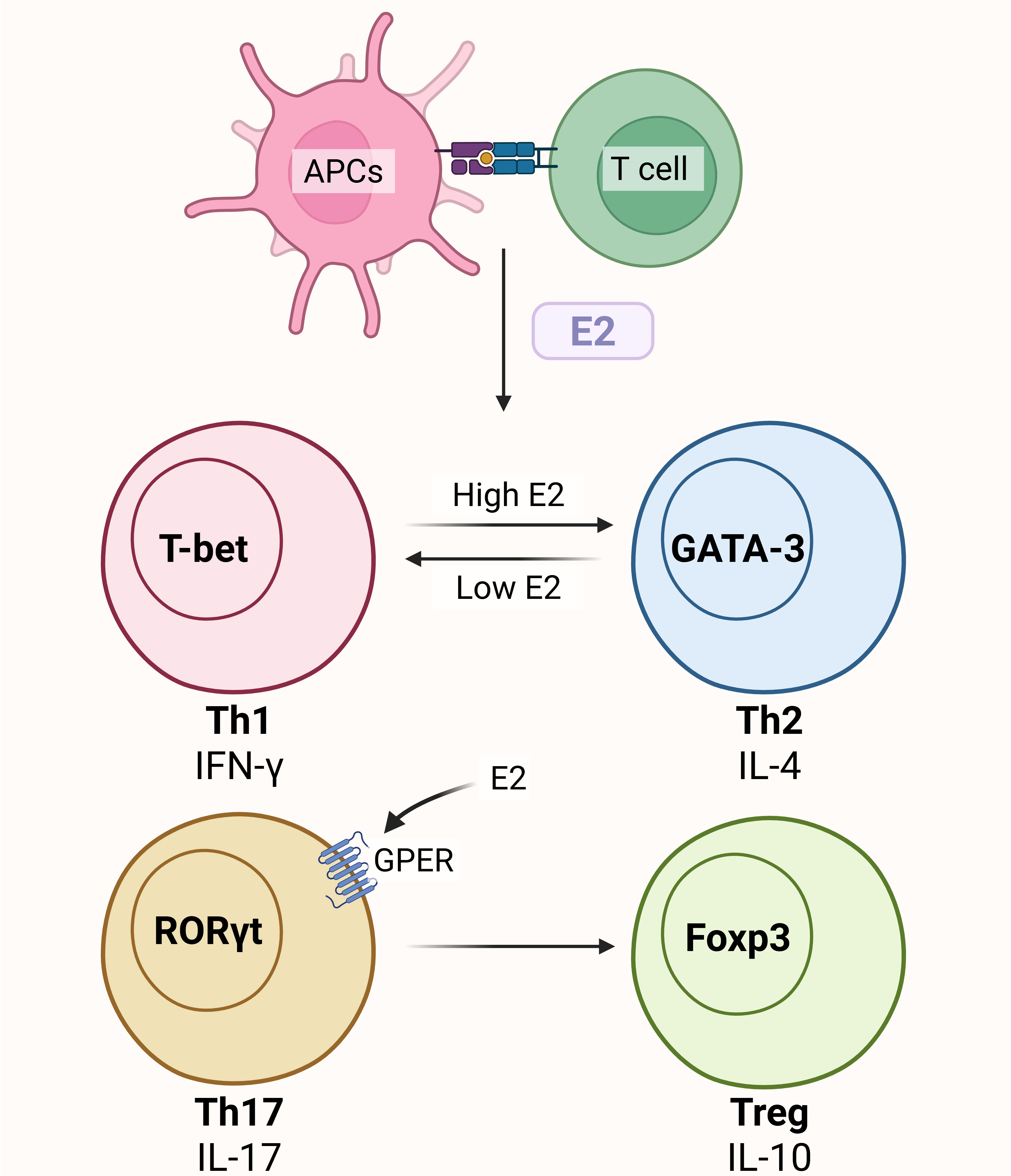
Figure 3. Estrogen regulates T cell phenotype conversion. Treg: Regulatory T cells; APCs: antigen-presenting cells; IFN-γ: interferon gamma; IL-17: interleukin-17; IL-10: interleukin-10; GPER: G protein-coupled estrogen receptor; GATA-3: GATA binding protein 3. The figure was created with https://app.biorender.com/illustrations/68b8e0321be53d1e5f76bfb2.
Female mice were found to have more autoimmune tendencies (higher amounts of germinal center B cells and autoantibodies) than male mice and to rely more on B1 cells secreting native IgM for
Estrogen has complicated effects on vascular inflammatory responses that vary depending on age, cell type, and hormone levels. Estradiol, for instance, reduces the inflammatory response to C-reactive protein in young (10 weeks) mice’s vascular smooth muscle cells and bone marrow-derived macrophages, but this effect is not present in aged (52 weeks) mice[106]. This implies that the preventive benefits of estrogen on the cardiovascular system may be diminished with age. How long may the beneficial effects of estrogen last as people age? Is estrogen replacement treatment a feasible option for postmenopausal women, considering the sharp increase in their risk of ASCVD? These fundamental questions are still open.
The benefits of estrogen on cardiovascular disease were noted in several studies starting in the 1980s. These findings led to the usage of estrogen therapy, which peaked in the late 1990s[107]. Estrogen treatment did not improve the primary or secondary prevention of cardiovascular events linked to coronary heart disease, according to two later randomized trials. In fact, it increased the risk of cardiovascular events[108-110]. Subsequent research investigated the subject further and found that women who began estrogen therapy less than ten years after menopause experienced fewer cardiac events and lower mortality than those who began more than ten years later. This discovery led to the “time hypothesis”, which postulates that estrogen slows the progression of cardiovascular disease but does not reverse pre-existing vascular damage. Although beginning estrogen medication during menopause decreases the development of ASCVD in women with healthy arteries, the effects of estrogen wane as vascular impairment arises after a certain age. The lower effectiveness of estrogen therapy may also be caused by biological variations between endogenous estradiol and synthetic hormones.
Additionally, recent research has put forth a novel theory: postmenopausal women may not gain as much from HRT due to age-related iron buildup[111]. This study discovered that postmenopausal women’s iron overload in plaques causes ERα proteolysis, which is exacerbated by the presence of estrogen and reverses the anti-atherosclerotic benefits of estrogen. This mechanism is intimately linked to the ubiquitination and degradation of ERα mediated by murine double minute-2 (Mdm2) under the impact of E2 and iron. The administration of iron chelators reduced the progression of atherosclerotic lesions caused by E2 and restored ERα expression levels. For postmenopausal women, combining HRT with iron restriction therapy may be a long-term way to stop the growth of atherosclerosis. Changes in iron metabolism status, separate from estrogen changes, are another significant factor determining AS risk in postmenopausal women, according to additional research findings[112]. In premenopausal women, menstruation is the main cause of iron loss, which may have an impact on cardiovascular metabolic risk factors. If confirmed, this fresh viewpoint has the potential to change how we think about gender disparities and offer fresh targets for cardiovascular and metabolic health interventions.
Women with late menopause (≥ 55 years) have a 10%-20% lower risk of cardiovascular diseases (CVD) than those with normal menopause (45-54 years), which is attributed to less oxidative stress from mitochondrial ROS (mitoROS)[113]. The NLRP3 inflammasome can be further activated by ROS. Large volumes of IL-1β and
Androgen
For men, testosterone is the most significant sex hormone since it may attach to androgen receptors in cells. Androgens have a two-way effect on the regulation of inflammation and immunity in atherosclerosis. Bidirectional regulatory effects of androgens can be attributed to the kind of target cell, the environment of action, and hormone levels.
Research has shown that androgens have anti-inflammatory effects on the activities of the immune system. Female mice that receive androgen treatment have T cells that produce more IL-10, indicating improved Th2 responses[114]. Atherosclerosis becomes more reliant on NLRP3 inflammasomes after castration, indicating that testosterone may have anti-atherosclerotic effects via reducing inflammation by blocking NLRP3 inflammasomes[115]. Testosterone inhibits the production of foam cells. In tiny pigs fed a high-fat, high-cholesterol diet, testosterone shortage causes an increase in the area of atherosclerotic lesions, the buildup of foam cells, and raised serum lipid levels[116].
Androgens may, however, have pro-inflammatory effects, according to some research. A higher risk of cardiovascular calcification has been linked to elevated testosterone levels[117]. Men have substantially higher levels of pro-atherosclerotic cytokines (such as IL-1β, IL-6, and TNF-α) than women do, and these cytokines are correlated with testosterone and the testosterone/estradiol ratio[76]. This implies that systemic inflammation may be linked to higher testosterone levels, raising the risk of atherosclerosis. The relationship between testosterone and inflammatory cytokines is still debatable, though. According to certain research, testosterone supplementation can increase the synthesis of IL-10 while decreasing TNFα, IL-1β, and IL-6[118]. The atherosclerosis context was not used to achieve these contentious results. It is necessary to clarify the general effects of testosterone on immunomodulation as well as its particular relevance in atherosclerosis.
The most frequent cause of hyperandrogenism in women of reproductive age is PCOS, and women with PCOS are known to be at a higher risk of developing ASCVD[119]. According to research, hyperandrogenism can increase the risk of insulin resistance and abnormal glucose and lipid metabolism in women, which can indirectly contribute to the onset and progression of ASCVD. It can also disrupt vascular endothelial function and promote atherosclerosis through the androgen receptor pathway.
Animal models
The estrogen-deficient state that follows female menopause is often simulated, and its consequences on atherosclerosis are studied using animal models, especially ovariectomized female mice or rats. Female mice and humans, however, differ significantly in that the former have shorter hormone/estrus cycles, lower levels of total hormones in the blood, and do not experience natural menopause[120].
While human lesions primarily occur in the coronary and carotid arteries, small animals such as mice (e.g., ApoE-/- and LDLR-/- models) acquire aortic root plaques. There are still variations in coronary plaque morphology even if the vascular anatomy of large animals (such as pigs and rabbits) more closely resembles that of humans[121].
Humans and animal models have different metabolisms as well. It takes a very high cholesterol diet (serum cholesterol > 1,000 mg/dL) in mice to cause plaques, which is significantly higher than the pathological levels in humans. Additionally, there are notable differences in the phenotype and composition of immune cells between mouse and human plaques. Complex immune cell interactions in human plaques result in certain mechanistic discoveries that are not as well-validated in humans. Nevertheless, the most common model for researching menopause is still ovariectomized mice.
Immunogenetic regulation of sex chromosomes
Available evidence indicates that genetic sex (XX or XY sex chromosomes) plays a significant role in sex differences in immune cell function[122]. The X chromosome is typically larger and more complex than the Y chromosome, and it contains a large number of genes associated with immune function. To maintain gene expression levels comparable to males (XY), females (XX) undergo random inactivation of one X chromosome through the mechanism of X chromosome inactivation (XCI) during early embryonic development. This prevents the simultaneous expression of both X chromosomes, which would cause an imbalance of genes within the cell. However, during XCI, approximately 15% of genes escape the silencing mechanism and maintain continuous expression. These genes are termed XCI escape genes, and many lack functional counterparts on the Y chromosome. Research indicates that XCI escape genes are enriched with numerous immune-related genes (such as TLR7, TLR8, CD40L, etc.). The biallelic expression of these genes may contribute to heightened immune responses in females, offering a potential mechanism for understanding the higher prevalence of autoimmune diseases among women[123].
Current research has confirmed the presence of active immune responses within atherosclerotic plaques, with notable gender differences in their development and progression. The X chromosome harbors numerous immune-regulatory genes. Therefore, whether X-linked mechanisms (such as XCI escape or skew) influence the gender disparities in atherosclerosis and the markedly elevated risk in postmenopausal women by modulating immune responses represents a critical scientific question for elucidating the
Among female immune cell subsets, the expression levels of XCI escape genes are typically significantly higher than in males. A study conducting quantitative analysis of XCI bias in 154 female atherosclerotic plaques and 55 blood samples found that XCI bias was prevalent in female carotid plaques (49.4%), significantly correlated with intraplaque hemorrhage, and capable of predicting the risk of peripheral arterial events within 3 years after carotid endarterectomy[124].
Previously, X inactivation-specific transcript (Xist) RNA was thought to primarily mediate XCI for gene dosage compensation. However, recent research evidence indicates that Xist participates in regulating broader gene expression networks through complex epigenetic mechanisms. Its role in the cardiovascular system is increasingly prominent. For instance, in atherosclerosis, Xist binds to various microRNAs (miRNAs), releasing miRNA suppression on their target genes. This affects the function of endothelial cells, smooth muscle cells, and macrophages, directly contributing to plaque formation and progression[125].
In summary, the dose advantage conferred by XCI escape in female X-linked immune genes is a double-edged sword. On the one hand, it enhances immune responses against infections; on the other hand, it increases susceptibility to autoimmune diseases and inflammation-driven ASCVD. This partially explains, from a genetic standpoint, the significant increase in ASCVD risk observed in postmenopausal women. Therefore, the marked rise in AS risk after menopause is not attributable to a single factor but rather results from the combined effects of genetic background (X chromosome) and altered hormonal environment (sharp decline in estrogen).
Other studies have simultaneously confirmed that Y-chromosomal genetic variations (such as the I1 haplotype) are associated with coronary artery disease risk, leading to altered UTY gene expression, regulating immune responses, and promoting atherosclerosis[126]. Clinical studies confirm that sex chromosome aneuploidies, such as Klinefelter syndrome (XXY), XYY syndrome, and Turner syndrome (XO), are frequently associated with abnormal sex hormone levels and metabolic disorders. These abnormalities further contribute to the development of a pro-atherogenic lipid profile, ultimately significantly increasing susceptibility to coronary artery disease[127].
However, the specific molecular mechanisms by which sex chromosome-associated mechanisms influence gender susceptibility to cardiovascular diseases such as atherosclerosis through differential regulation of immune responses remain a weak link in current research and warrant further investigation.
The establishment of the four-core genotype (FCG) mouse model has been instrumental in distinguishing chromosomal effects, gonadal effects, and their interactions. The Sry gene on the Y chromosome regulates testis development, while its absence leads to ovarian formation. Thus, XX chromosomes typically coexist with ovaries, while XY chromosomes correlate with testes, making it challenging to distinguish the specific roles between genetic sex and gonadal sex. The FCG model achieves this by translocating the testis-determining gene Sry from the Y chromosome to an autosome. This successfully separates the effects of gonadal hormones from those of sex chromosomes, yielding four genotype combinations: XX females with ovaries, XX males with testes, XY females with ovaries, and XY males with testes[128]. XX mice (regardless of sex) exhibited higher body weight, blood lipid levels, and more severe atherosclerotic lesions. Concurrently, the study revealed that the effect of gonadectomy on lesion area was chromosome-specific: ovariectomy only exacerbated lesions in XX mice, while orchiectomy only alleviated lesions in XY mice[129].
The XY* mouse model is used to distinguish between X- or Y-chromosome dosage effects. If the target trait is driven by the number of X chromosomes, mice carrying two X chromosomes (XX and XXY) will differ from those with only one X chromosome (XY and XO). If the target trait is determined by the presence or absence of the Y chromosome, mice lacking the Y chromosome (XX and XO) will contrast with those retaining it (XY and XXY).
LIMITATIONS
Although this study summarizes current knowledge on the regulatory roles of sex hormones and sex chromosomes in immune cells during female atherosclerosis, many questions remain unanswered. While the crucial role of the complement system in atherosclerosis is acknowledged and efforts have been made to understand its sex-specific activation, there is still a lack of direct experimental evidence. Future studies should clarify how the complement system contributes to sex-related disparities in atherosclerosis.
Studies including human female patients remain rare, although single-cell sequencing offers high-resolution data on immune cell heterogeneity within the plaque microenvironment. As a result, immune cell characteristics in atherosclerotic plaques across various age groups of women are not fully characterized.
We discussed the immunomodulatory role of estrogen in atherosclerosis, but further research is needed on how to optimize the timing, dosage, and administration of hormone therapy, as well as to explore how immunotherapy can be applied in treating atherosclerosis in women.
CONCLUSION
Women have distinct epidemiological characteristics and a distinct etiology of atherosclerosis, the primary pathological underpinning of cardiovascular disease. By comparing the notable changes in ASCVD risk in pre- and postmenopausal women, several studies have validated the crucial function of estrogen in cardiovascular protection (including enhancement of lipid metabolism and promotion of M2 macrophage polarization).
In addition to the regulating effect of estrogen, women’s immune systems are distinct due to the danger of vascular injury linked to a high incidence of autoimmune illnesses and gender differences in immune cell activity (e.g., macrophages, T cells, and B cells). These characteristics collectively form a distinct pathophysiological network of atherosclerosis in women, offering fresh opportunities and avenues for the creation of gender-specific preventative and therapeutic approaches.
The Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) and the Colchicine Cardiovascular Outcomes Trial (COLCOT) have both shown that immunomodulation can successfully reduce major adverse cardiovascular events (MACEs) in recent years[130,131]. This supports the effectiveness of immunomodulation in the management of atherosclerosis and offers crucial information for investigating immunotherapy approaches tailored to women.
The gender-specific immunological processes of atherosclerosis in women have not been fully explored, and early epidemiological studies and therapeutic trials have long been male-dominated, with a higher proportion of male samples included. Future research on the immunological mechanism behind atherosclerosis in women of all ages should be improved, and strategies for utilizing the unique immune traits of women to optimize atherosclerosis prevention and treatment should be investigated.
DECLARATIONS
Acknowledgments
We gratefully acknowledge the support from the National Natural Science Foundation of China (Nos. 82470341, 62135002, U24 A20648), the Key Research and Development Program of Heilongjiang Provincial Science and Technology Department (2024ZX12C01), and the Natural Science Foundation of Heilongjiang Provincial Science and Technology Department (PL2024H085). Figures were created with BioRender.com.
Authors’ contributions
Designed the framework and direction of the manuscript: Wei X, Li M, Cui Z, Hu S,
Wrote the first draft of the manuscript: Wei X, Li M, Cui Z
Produced the first draft of illustration: Wei X, Li M, Xu B
Funding acquisition: Hu S, Yu B, Jia H
Made critical revision of the manuscript: Wei X, Li M, Cui Z, Wang W, Shao S, Yi B, Xu B, Ren H, Jia H, Yu B, Hu S
All authors approved the final manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
Dr. Yu B received support from the National Natural Science Foundation of China (62135002). Dr. Jia H received support from the National Natural Science Foundation of China (No. U24 A20648). Dr. Hu S received support from the National Natural Science Foundation of China (82470341), Key Research and Development Program of Heilongjiang Provincial Science and Technology Department (2024ZX12C01), and Fund of the Natural Science Foundation of Heilongjiang Provincial Science and Technology Department (PL2024H085). This work was supported by the above funds.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1204-22.
2. Chong B, Jayabaskaran J, Jauhari SM, et al. Global burden of cardiovascular diseases: projections from 2025 to 2050. Eur J Prev Cardiol. 2025;32:1001-15.
3. El Khoudary SR, Aggarwal B, Beckie TM, et al.; American Heart Association Prevention Science Committee of the Council on Epidemiology and Prevention; and Council on Cardiovascular and Stroke Nursing. Menopause transition and cardiovascular disease risk: implications for timing of early prevention: a scientific statement from the American Heart Association. Circulation 2020;142:e506-32.
4. Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131-8.
5. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709-21.
6. Tabas I, Lichtman AH. Monocyte-macrophages and T cells in atherosclerosis. Immunity. 2017;47:621-34.
7. Fang F, Xiao C, Li C, Liu X, Li S. Tuning macrophages for atherosclerosis treatment. Regen Biomater. 2023;10:rbac103.
8. Zernecke A, Winkels H, Cochain C, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020;127:402-26.
9. Wang Y, Wang Q, Xu D. New insights into macrophage subsets in atherosclerosis. J Mol Med. 2022;100:1239-51.
10. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. 2022;22:251-65.
11. Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. 2018;9:113.
12. Evrard M, Kwok IWH, Chong SZ, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity. 2018;48:364-379.e8.
13. Luo J, Thomassen JQ, Nordestgaard BG, Tybjærg-Hansen A, Frikke-Schmidt R. Neutrophil counts and cardiovascular disease. Eur Heart J. 2023;44:4953-64.
14. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837-45.
15. Ma Y, Yabluchanskiy A, Iyer RP, et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110:51-61.
16. Qi H, Yang S, Zhang L. Neutrophil extracellular traps and endothelial dysfunction in atherosclerosis and thrombosis. Front Immunol. 2017;8:928.
17. Yang X, Ma Y, Chen X, Zhu J, Xue W, Ning K. Mechanisms of neutrophil extracellular trap in chronic inflammation of endothelium in atherosclerosis. Life Sci. 2023;328:121867.
18. Döring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res. 2017;120:736-43.
19. Gomez I, Ward B, Souilhol C, et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat Commun. 2020;11:214.
20. Tiberio L, Del Prete A, Schioppa T, Sozio F, Bosisio D, Sozzani S. Chemokine and chemotactic signals in dendritic cell migration. Cell Mol Immunol. 2018;15:346-52.
21. Sun Y, Zhou L, Chen W, et al. Immune metabolism: a bridge of dendritic cells function. Int Rev Immunol. 2022;41:313-25.
22. Qian C, Cao X. Dendritic cells in the regulation of immunity and inflammation. Semin Immunol. 2018;35:3-11.
23. Kiss MG, Binder CJ. The multifaceted impact of complement on atherosclerosis. Atherosclerosis. 2022;351:29-40.
24. Speidl WS, Kastl SP, Huber K, Wojta J. Complement in atherosclerosis: friend or foe? J Thromb Haemost. 2011;9:428-40.
25. Si W, He P, Wang Y, et al. Complement complex C5b-9 levels are associated with the clinical outcomes of acute ischemic stroke and carotid plaque stability. Transl Stroke Res. 2019;10:279-86.
26. Bhatia VK, Yun S, Leung V, et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol. 2007;170:416-26.
27. Spivia W, Magno PS, Le P, Fraser DA. Complement protein C1q promotes macrophage anti-inflammatory M2-like polarization during the clearance of atherogenic lipoproteins. Inflamm Res. 2014;63:885-93.
28. Fraser DA, Tenner AJ. Innate immune proteins C1q and mannan-binding lectin enhance clearance of atherogenic lipoproteins by human monocytes and macrophages. J Immunol. 2010;185:3932-9.
29. Maffia P, Mauro C, Case A, Kemper C. Canonical and non-canonical roles of complement in atherosclerosis. Nat Rev Cardiol. 2024;21:743-61.
30. Monaco C, Dib L. Atheroimmunology: keeping the immune system in atherosclerosis in check. Nat Rev Cardiol. 2024;21:737-8.
31. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17:387-401.
32. Chang S, Wang Z, An T. T-cell metabolic reprogramming in atherosclerosis. Biomedicines. 2024;12:1844.
33. Fernandez DM, Rahman AH, Fernandez NF, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576-88.
34. Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454-60.
35. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. 2019;50:892-906.
36. Gisterå A, Robertson AK, Andersson J, et al. Transforming growth factor-β signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci Transl Med. 2013;5:196ra100.
37. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. 2015;35:280-7.
38. Baardman J, Lutgens E. Regulatory T cell metabolism in atherosclerosis. Metabolites. 2020;10:279.
39. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity. 2013;39:949-62.
40. Ryu H, Lim H, Choi G, et al. Atherogenic dyslipidemia promotes autoimmune follicular helper T cell responses via IL-27. Nat Immunol. 2018;19:583-93.
41. Gaddis DE, Padgett LE, Wu R, et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun. 2018;9:1095.
42. Poznyak AV, Bezsonov EE, Popkova TV, Starodubova AV, Orekhov AN. Immunity in atherosclerosis: focusing on T and B cells. Int J Mol Sci. 2021;22:8379.
43. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. 2019;16:180-96.
44. Ylä-Herttuala S, Palinski W, Butler SW, Picard S, Steinberg D, Witztum JL. Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler Thromb. 1994;14:32-40.
45. Kyaw T, Tipping P, Bobik A, Toh BH. Opposing roles of B lymphocyte subsets in atherosclerosis. Autoimmunity. 2017;50:52-6.
46. Baumgarth N. B-1 Cell Heterogeneity and the regulation of natural and antigen-induced IgM production. Front Immunol. 2016;7:324.
47. Abelman R, Tien PC. The reproductive transition: effects on viral replication, immune activation, and metabolism in women with HIV infection. Curr HIV/AIDS Rep. 2022;19:133-9.
48. Ahmed S, Spence JD. Sex differences in the intestinal microbiome: interactions with risk factors for atherosclerosis and cardiovascular disease. Biol Sex Differ. 2021;12:35.
49. Barcena ML, Christiansen-Mensch C, Aslam M, Haritonow N, Ladilov Y, Regitz-Zagrosek V. Upregulation of mitochondrial Sirt3 and alleviation of the inflammatory phenotype in macrophages by estrogen. Cells. 2024;13:1420.
50. Rosenzweig R, Gupta S, Kumar V, Gumina RJ, Bansal SS. Estrogenic bias in T-Lymphocyte biology: Implications for cardiovascular disease. Pharmacol Res. 2021;170:105606.
51. Yasrebi A, Regan D, Roepke TA. The influence of estrogen response element ERα signaling in the control of feeding behaviors in male and female mice. Steroids. 2023;195:109228.
52. Smit V, de Mol J, Kleijn MNAB, et al. Sexual dimorphism in atherosclerotic plaques of aged Ldlr-/- mice. Immun Ageing. 2024;21:27.
53. Chaithra RJ, Fremingston M, Bharathi P. Coronary artery atherosclerosis in reproductive age group women-an autopsy study. Asian J Med Sci. 2024;15:145-50.
54. Centner A, Ukhanov V, Laitano O, Hill S, Cullen A, Salazar G. Sex differences in atherosclerosis in ApoE-/- mice exposed to nicotine and cigarette smoke. FASEB J. 2021:35.
55. Gallucci G, Tartarone A, Lerose R, Lalinga AV, Capobianco AM. Cardiovascular risk of smoking and benefits of smoking cessation. J Thorac Dis. 2020;12:3866-76.
56. Connelly PJ, Currie G, Delles C. Sex differences in the prevalence, outcomes and management of hypertension. Curr Hypertens Rep. 2022;24:185-92.
57. Medina D, Mehay D, Arnold AC. Sex differences in cardiovascular actions of the renin-angiotensin system. Clin Auton Res. 2020;30:393-408.
58. Park J, Son MK, Park HY. Substantial lipid increases during menopausal transition in Korean middle-aged women. J Korean Med Sci. 2023;38:e238.
59. Meng Q, Chao Y, Zhang S, et al. Attenuation of estrogen and its receptors in the post-menopausal stage exacerbates dyslipidemia and leads to cognitive impairment. Mol Brain. 2023;16:80.
60. El Khoudary SR, Chen X, Wang Z, et al. Low-density lipoprotein subclasses over the menopausal transition and risk of coronary calcification and carotid atherosclerosis: the SWAN heart and HDL ancillary studies. Menopause. 2023;30:1006-13.
61. Fernández-Suárez ME, Escolà-Gil JC, Pastor O, et al. Clinically used selective estrogen receptor modulators affect different steps of macrophage-specific reverse cholesterol transport. Sci Rep. 2016;6:32105.
62. Mauvais-Jarvis F. Sex differences in energy metabolism: natural selection, mechanisms and consequences. Nat Rev Nephrol. 2024;20:56-69.
63. Bjune JI, Strømland PP, Jersin RÅ, Mellgren G, Dankel SN. Metabolic and epigenetic regulation by estrogen in adipocytes. Front Endocrinol. 2022;13:828780.
64. Klaver M, de Blok CJM, Wiepjes CM, et al. Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. Eur J Endocrinol. 2018;178:163-71.
65. Mauvais-Jarvis F, Manson JE, Stevenson JC, Fonseca VA. Menopausal hormone therapy and type 2 diabetes prevention: evidence, mechanisms, and clinical implications. Endocr Rev. 2017;38:173-88.
66. Juutilainen A, Kortelainen S, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Gender difference in the impact of type 2 diabetes on coronary heart disease risk. Diabetes Care. 2004;27:2898-904.
68. Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham study. Am J Epidemiol. 1997;145:408-15.
69. Mortensen MB, Jensen JM, Rønnow Sand NP, et al. Association of autoimmune diseases with coronary atherosclerosis severity and ischemic events. J Am Coll Cardiol. 2024;83:2643-54.
70. Yuan XM, Ward LJ, Forssell C, Siraj N, Li W. Carotid atheroma from men has significantly higher levels of inflammation and iron metabolism enabled by macrophages. Stroke. 2018;49:419-25.
71. Yahagi K, Davis HR, Arbustini E, Virmani R. Sex differences in coronary artery disease: pathological observations. Atherosclerosis. 2015;239:260-7.
72. Partida RA, Libby P, Crea F, Jang IK. Plaque erosion: a new in vivo diagnosis and a potential major shift in the management of patients with acute coronary syndromes. Eur Heart J. 2018;39:2070-6.
73. Huang Z, Chen B, Liu X, et al. Effects of sex and aging on the immune cell landscape as assessed by single-cell transcriptomic analysis. Proc Natl Acad Sci U S A. 2021:118.
74. Sukhavasi K, Mocci G, Ma L, et al. Single-cell RNA sequencing reveals sex differences in the subcellular composition and associated gene-regulatory network activity of human carotid plaques. Nat Cardiovasc Res. 2025;4:412-32.
75. Saigusa R, Vallejo J, Gulati R, et al. Sex differences in coronary artery disease and diabetes revealed by scRNA-Seq and CITE-Seq of human CD4+ T Cells. Int J Mol Sci. 2022;23:9875.
76. Bernardi S, Toffoli B, Tonon F, et al. Sex differences in proatherogenic cytokine levels. Int J Mol Sci. 2020;21:3861.
77. Yerly A, van der Vorst EPC, Baumgartner I, Bernhard SM, Schindewolf M, Döring Y. Sex-specific and hormone-related differences in vascular remodelling in atherosclerosis. Eur J Clin Invest. 2023;53:e13885.
78. Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor α variant (ER46) in human endothelial cells. Proc Natl Acad Sci U S A. 2003;100:4807-12.
79. Li G, Chen YF, Kelpke SS, Oparil S, Thompson JA. Estrogen attenuates integrin-β3-dependent adventitial fibroblast migration after inhibition of osteopontin production in vascular smooth muscle cells. Circulation. 2000;101:2949-55.
80. Meng Q, Li Y, Ji T, et al. Estrogen prevent atherosclerosis by attenuating endothelial cell pyroptosis via activation of estrogen receptor α-mediated autophagy. J Adv Res. 2021;28:149-64.
81. Liang D, Feng Y, Zandkarimi F, et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186:2748-2764.e22.
82. Folahan JT, Olorundare OE, Ajayi AM, et al. Oxidized dietary lipids induce vascular inflammation and atherogenesis in post-menopausal rats: estradiol and selected antihyperlipidemic drugs restore vascular health in vivo. Lipids Health Dis. 2023;22:107.
83. Lynch S, Boyett JE, Smith MR, Giordano-Mooga S. Sex hormone regulation of proteins modulating mitochondrial metabolism, dynamics and inter-organellar cross talk in cardiovascular disease. Front Cell Dev Biol. 2020;8:610516.
84. Miller AP, Feng W, Xing D, et al. Estrogen modulates inflammatory mediator expression and neutrophil chemotaxis in injured arteries. Circulation. 2004;110:1664-9.
85. Evans BR, Yerly A, van der Vorst EPC, et al. Inflammatory mediators in atherosclerotic vascular remodeling. Front Cardiovasc Med. 2022;9:868934.
86. McCrohon JA, Nakhla S, Jessup W, Stanley KK, Celermajer DS. Estrogen and progesterone reduce lipid accumulation in human monocyte-derived macrophages: a sex-specific effect. Circulation. 1999;100:2319-25.
87. Bao Z, Liu ZQ, He PY, Adali J, Yang YC, Wulasihan M. 17β-estradiol regulates adenosine triphosphate-binding cassette transporters A1 expression via estrogen receptor A to increase macrophage cholesterol efflux. J Physiol Pharmacol. 2023:74.
88. Keselman A, Fang X, White PB, Heller NM. Estrogen signaling contributes to sex differences in macrophage polarization during asthma. J Immunol. 2017;199:1573-83.
89. Toniolo A, Fadini GP, Tedesco S, et al. Alternative activation of human macrophages is rescued by estrogen treatment in vitro and impaired by menopausal status. J Clin Endocrinol Metab. 2015;100:E50-8.
90. Murphy AJ, Guyre PM, Pioli PA. Estradiol suppresses NF-kappa B activation through coordinated regulation of let-7a and miR-125b in primary human macrophages. J Immunol. 2010;184:5029-37.
91. Ghisletti S, Meda C, Maggi A, Vegeto E. 17β-estradiol inhibits inflammatory gene expression by controlling NF-κB intracellular localization. Mol Cell Biol. 2005;25:2957-68.
92. Polari L, Wiklund A, Sousa S, et al. SERMs Promote anti-inflammatory signaling and phenotype of CD14+ cells. Inflammation. 2018;41:1157-71.
93. Chen Y, Zhao H, Ren X. Estrogen and progestogen inhibit NF-κB in atherosclerotic tissues of ovariectomized ApoE-/- mice. Climacteric. 2016;19:357-63.
94. Meng Q, Bi Y, Feng H, et al. Activation of estrogen receptor α inhibits TLR4 signaling in macrophages and alleviates the instability of atherosclerotic plaques in the postmenopausal stage. Int Immunopharmacol. 2023;116:109825.
95. Liu SL, Bajpai A, Hawthorne EA, et al. Cardiovascular protection in females linked to estrogen-dependent inhibition of arterial stiffening and macrophage MMP12. JCI Insight. 2019:4.
96. Marczell I, Hrabak A, Nyiro G, et al. 17-β-estradiol decreases neutrophil superoxide production through Rac1. Exp Clin Endocrinol Diabetes. 2016;124:588-92.
97. Zhang P, Fu Y, Ju J, et al. Estradiol inhibits fMLP-induced neutrophil migration and superoxide production by upregulating MKP-2 and dephosphorylating ERK. Int Immunopharmacol. 2019;75:105787.
98. Yasuda H, Sonoda A, Yamamoto M, et al. 17-β-estradiol enhances neutrophil extracellular trap formation by interaction with estrogen membrane receptor. Arch Biochem Biophys. 2019;663:64-70.
99. Murakami H, Ishikawa M, Higashi H, et al. Equol, a soybean metabolite with estrogen-like functions, decreases lipopolysaccharide-induced human neutrophil extracellular traps in vitro. Shock. 2024;61:695-704.
100. Yao Y, Cai X, Chen Y, Zhang M, Zheng C. Estrogen deficiency-mediated osteoimmunity in postmenopausal osteoporosis. Med Res Rev. 2025;45:561-75.
101. Notas G, Kampa M, Castanas E. G protein-coupled estrogen receptor in immune cells and its role in immune-related diseases. Front Endocrinol. 2020;11:579420.
102. Brunsing RL, Owens KS, Prossnitz ER. The G protein-coupled estrogen receptor (GPER) agonist G-1 expands the regulatory T-cell population under TH17-polarizing conditions. J Immunother. 2013;36:190-6.
103. Salem ML. Estrogen, a double-edged sword: modulation of TH1- and TH2-mediated inflammations by differential regulation of TH1/TH2 cytokine production. Curr Drug Targets Inflamm Allergy. 2004;3:97-104.
104. Alanazi H, Zhang Y, Fatunbi J, Luu T, Kwak-Kim J. The impact of reproductive hormones on T cell immunity; normal and assisted reproductive cycles. J Reprod Immunol. 2024;165:104295.
105. Bagchi-Chakraborty J, Francis A, Bray T, et al. B cell Fcγ receptor IIb modulates atherosclerosis in male and female mice by controlling adaptive germinal center and innate B-1-cell responses. Arterioscler Thromb Vasc Biol. 2019;39:1379-89.
106. Bowling MR, Xing D, Kapadia A, et al. Estrogen effects on vascular inflammation are age dependent: role of estrogen receptors. Arterioscler Thromb Vasc Biol. 2014;34:1477-85.
107. Cho L, Kaunitz AM, Faubion SS, et al.; ACC CVD in women committee. rethinking menopausal hormone therapy: for whom, what, when, and how long? Circulation 2023;147:597-610.
108. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-13.
109. Manson JE, Hsia J, Johnson KC, et al.; Women’s Health Initiative Investigators. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med 2003;349:523-34.
110. Anderson GL, Limacher M, Assaf AR, et al. ; Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701-12.
111. Xu T, Cai J, Wang L, et al. Hormone replacement therapy for postmenopausal atherosclerosis is offset by late age iron deposition. Elife. 2023:12.
112. Ahanchi NS, Khatami F, Llanaj E, et al. The complementary roles of iron and estrogen in menopausal differences in cardiometabolic outcomes. Clin Nutr. 2024;43:1136-50.
113. Darvish S, Murray KO, Ludwig KR, et al. Preservation of vascular endothelial function in late-onset postmenopausal women. Circ Res. 2025;136:455-69.
114. Nasser SA, Afify EA, Kobeissy F, Hamam B, Eid AH, El-Mas MM. Inflammatory basis of atherosclerosis: modulation by sex hormones. Curr Pharm Des. 2021;27:2099-111.
115. Chen S, Markman JL, Shimada K, et al. Sex-specific effects of the Nlrp3 inflammasome on atherogenesis in LDL receptor-deficient mice. JACC Basic Transl Sci. 2020;5:582-98.
116. Deng L, Fu D, Zhu L, Huang J, Ling Y, Cai Z. Testosterone deficiency accelerates early stage atherosclerosis in miniature pigs fed a high-fat and high-cholesterol diet: urine 1H NMR metabolomics targeted analysis. Mol Cell Biochem. 2021;476:1245-55.
117. Woodward HJ, Zhu D, Hadoke PWF, MacRae VE. Regulatory role of sex hormones in cardiovascular calcification. Int J Mol Sci. 2021;22:4620.
118. Malkin CJ, Pugh PJ, Jones RD, Kapoor D, Channer KS, Jones TH. The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J Clin Endocrinol Metab. 2004;89:3313-8.
119. Mani H, Levy MJ, Davies MJ, et al. Diabetes and cardiovascular events in women with polycystic ovary syndrome: a 20-year retrospective cohort study. Clin Endocrinol. 2013;78:926-34.
120. Fontaine C, Gosset A, Davezac M, et al. From sex hormone decrease to hormonal treatment: impacts on cardiovascular risk with ageing. Cardiovasc Res. 2025;121:1551-65.
121. Man JJ, Beckman JA, Jaffe IZ. Sex as a biological variable in atherosclerosis. Circ Res. 2020;126:1297-319.
122. Dunn SE, Perry WA, Klein SL. Mechanisms and consequences of sex differences in immune responses. Nat Rev Nephrol. 2024;20:37-55.
123. Feng Z, Liao M, Zhang L. Sex differences in disease: sex chromosome and immunity. J Transl Med. 2024;22:1150.
124. Buono MF, Benavente ED, Daniels M, et al. X chromosome inactivation skewing is common in advanced carotid atherosclerotic lesions in females and predicts secondary peripheral artery events. Biol Sex Differ. 2023;14:43.
125. Almalki WH. Unraveling the role of Xist RNA in cardiovascular pathogenesis. Pathol Res Pract. 2024;253:154944.
126. Eales JM, Maan AA, Xu X, et al. Human Y chromosome exerts pleiotropic effects on susceptibility to atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39:2386-401.
127. Arnold AP, Cassis LA, Eghbali M, Reue K, Sandberg K. Sex hormones and sex chromosomes cause sex differences in the development of cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2017;37:746-56.
128. Reue K, Wiese CB. Illuminating the mechanisms underlying sex differences in cardiovascular disease. Circ Res. 2022;130:1747-62.
129. AlSiraj Y, Chen X, Thatcher SE, et al. XX sex chromosome complement promotes atherosclerosis in mice. Nat Commun. 2019;10:2631.
130. Ridker PM, Everett BM, Thuren T, et al.; CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119-31.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.