


The role of microRNAs in acute and chronic heart failure: a key regulator and therapeutic target
 0
0 Abstract
Heart Failure (HF) represents the terminal stage of various cardiac diseases and is classified into Acute Heart Failure (AHF) and Chronic Heart Failure (CHF) based on the onset speed. Both are characterized by impaired myocardial function, neurohumoral disturbance, and ventricular remodeling, yet exhibit significant differences in pathophysiological mechanisms and clinical phenotypes. MicroRNAs (miRNAs), a class of non-coding RNAs approximately 18-25 nucleotides in length, regulate gene expression at the post-transcriptional level by targeting the 3' untranslated region (3'UTR) of target gene messenger RNAs. They are extensively involved in physiological and pathological processes such as cell proliferation, apoptosis, and fibrosis. A growing body of evidence has confirmed that abnormal miRNA expression profiles are closely associated with the occurrence and progression of HF, playing a crucial regulatory role in the pathological processes of both AHF and CHF. This review systematically examines the biological characteristics of miRNAs and elaborates on their core regulatory roles in the pathophysiological mechanisms of AHF and CHF. It analyzes differences and commonalities in miRNA expression and function between the two conditions, and explores the clinical application prospects of miRNAs as diagnostic markers, prognostic indicators, and therapeutic targets for HF. Finally, it summarizes current research challenges and future directions, aiming to provide a theoretical basis for the precise diagnosis and treatment of HF.
Keywords
INTRODUCTION
Heart Failure (HF) is a complex clinical syndrome characterized by impaired cardiac pumping function, resulting in an insufficient cardiac output to meet the body’s metabolic demands or the ability to maintain pumping function only when ventricular filling pressure is abnormally elevated[1]. As the end-stage manifestation of various cardiovascular diseases, HF is associated with persistently high morbidity and mortality rates. It not only severely compromises patients’ quality of life but also imposes a substantial burden on families and the broader healthcare system[2,3].
Based on clinical phenotype and disease progression, HF is mainly classified into two types: Acute Heart Failure (AHF) and Chronic Heart Failure (CHF)[4]. These two subtypes exhibit significant differences yet close correlations in their pathophysiological mechanisms. AHF is defined by acute cardiac decompensation, typically triggered by acute etiologies such as acute myocardial infarction (AMI), hypertensive crisis and acute myocarditis. It presents with sudden onset of impaired myocardial contractility, circulatory congestion and insufficient tissue perfusion, carrying extremely high mortality without timely intervention[5]. In contrast, CHF typically develops from the progression of chronic cardiovascular diseases including coronary artery disease, hypertensive heart disease, and cardiomyopathy. Its core pathological process is ventricular remodeling, accompanied by the sustained overactivation of the Renin-Angiotensin-Aldosterone System (RAAS) and Sympathetic Nervous System (SNS). This in turn induces a series of pathological changes such as myocardial hypertrophy, fibrosis, and electrical activity disorders, ultimately leading to the progressive deterioration of cardiac function[6]. Therefore, in-depth clarification of the core pathological mechanisms of HF and identification of novel targets with both diagnostic value and therapeutic potential have become urgent needs in the field of cardiovascular research.
MicroRNAs (miRNAs) are a class of endogenous non-coding RNAs approximately 22 nucleotides in length. Since their first discovery in Caenorhabditis elegans in 1993, their core role in gene expression regulation has been gradually elucidated, establishing them as a key focus in cardiovascular disease research[7]. miRNAs possess high tissue-specificity, cell-specificity, and spatiotemporal expression specificity. By binding to the 3' untranslated region (3'UTR) of target gene messenger RNAs (mRNAs), they regulate gene expression at the post-transcriptional level and participate in the construction of complex gene regulatory networks[8-10]. In the cardiovascular system, miRNAs are extensively involved in key physiological processes such as myocardial cell proliferation[11], differentiation[12], apoptosis[13], angiogenesis[14], and myocardial fibrosis[15], playing an important regulatory role in the development and homeostasis maintenance of the cardiovascular system. Given their important functions, aberrant miRNA expression has been confirmed to be closely associated with multiple core pathological processes in HF[16-18]. Notably, the intrinsic differences between the pathological responses of acute stress-induced AHF and chronic progressive pathological remodeling profoundly affect the expression profiles of miRNAs, regulatory preferences of target genes, and their functional roles in the two states of HF[19,20]. Accumulating evidence has confirmed that aberrant miRNA expression is closely associated with the occurrence and progression of both AHF and CHF, making miRNAs promising candidates for deciphering HF pathogenesis and developing precision therapies[21,22]. Thus, in-depth exploration of the role of miRNAs in HF not only contributes to unraveling the pathological essence of AHF and CHF but also provides a new theoretical basis for improving clinical diagnosis, prognosis evaluation, and therapeutic efficacy.
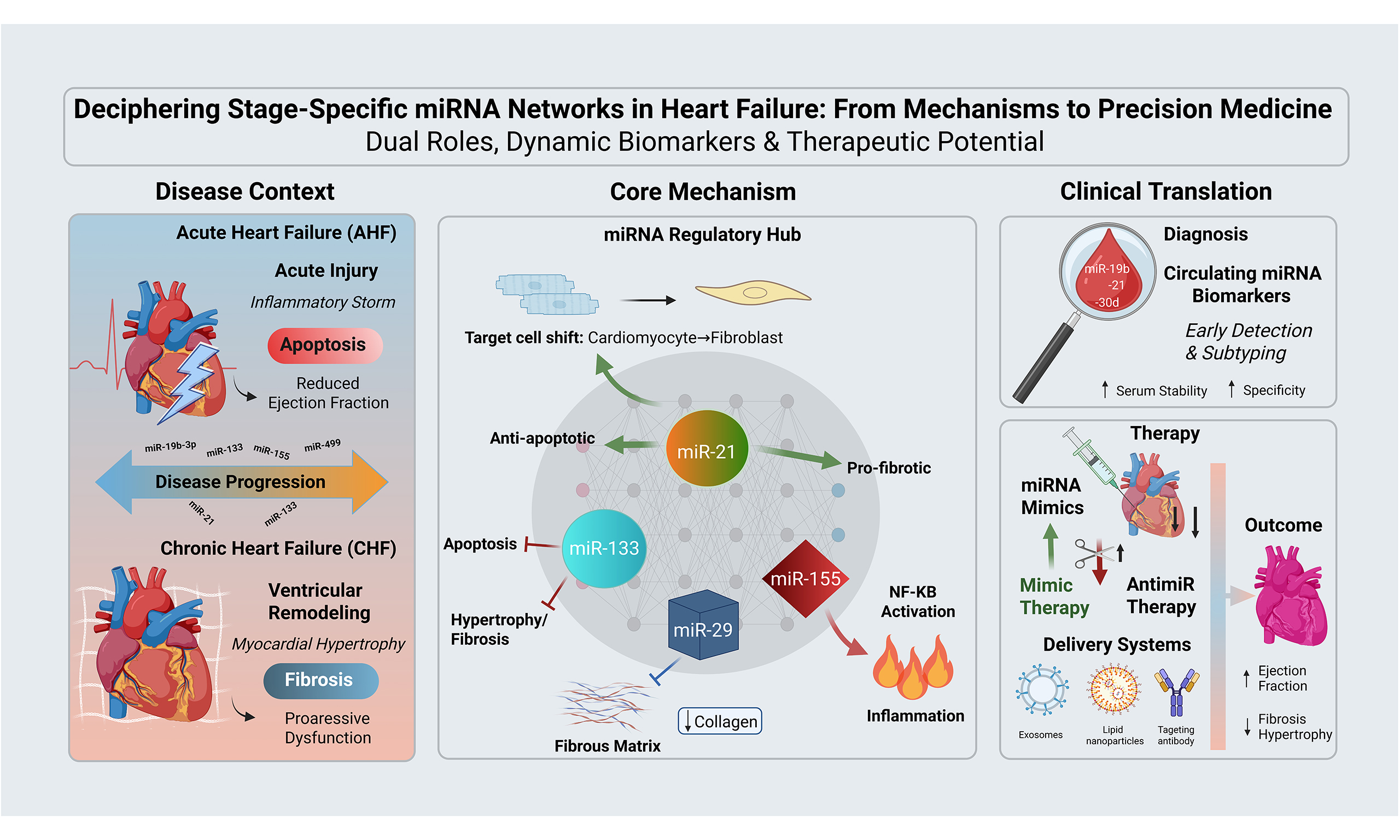
Behavioral differences of miRNAs between AHF and CHF
Temporal dynamics: acute stress response vs. chronic adaptive remodeling
miRNAs exhibit distinctly different temporal patterns of expression and function in AHF and CHF, mirroring the disease progression from initial injury to long-term structural alterations. In AHF, miRNA levels undergo rapid and robust dynamic changes during hospitalization. A study monitoring circulating miRNA levels in AHF patients at admission, 48-72 h post-admission, and 7-21 days later identified a significant decrease in miR-140-3p, a miRNA associated with mitochondrial fission, during treatment[23]. Such rapid fluctuations over days to weeks suggest miRNAs are involved in the body’s immediate response to acute hemodynamic collapse and cellular stress. Notably, low miR-140-3p levels correlate with higher long-term mortality and heart failure event rates, indicating that miRNA response patterns in the acute phase may determine long-term prognosis[23]. In contrast, CHF is characterized by miRNA expression establishing a relatively stable yet pathological set point, a pattern that aligns with long-term cardiac structural remodeling and dysfunction. For example, in a mouse model of pressure overload-induced heart failure, myocardial miR-152 expression is persistently upregulated, directly contributing to mitochondrial dysfunction and impaired contractile function[24]. This sustained abnormal expression drives chronic pathological progression.
Stress response biology: core pathophysiological focus across different stages
AHF and CHF exist in distinct pathophysiological states, leading their key miRNA regulatory networks to converge on different biological processes. The acute onset of AHF triggers severe energy metabolic crisis and cellular damage, prompting miRNA regulation to rapidly shift toward pathways involved in mitochondrial dynamics and acute cell apoptosis. Studies confirm that circulating miR-140-5p levels in AHF patients are closely associated with excessive mitochondrial fission in cardiomyocytes, a hallmark of disrupted energy metabolism and cell death[25]. Similarly, in AHF-related ischemic hepatitis, upregulated miR-17 correlates with mitochondrial dynamin protein dysregulation, reflecting a systemic multi-organ stress response to acute hypoperfusion[26]. These findings highlight that miRNA behavior in AHF centers on countering acute energy metabolic disorders and organelle dysfunction. The core pathological feature of CHF is progressive ventricular remodeling, with myocardial fibrosis and chronic low-grade inflammation as key drivers. Consequently, numerous critical miRNAs in CHF regulate these processes. miR-21 and the miR-29 family have been identified as core regulators of the inflammatory-fibrotic axis in heart failure with preserved ejection fraction (HFpEF), is known as a common form of CHF[27].
Cell type specificity
The dominant cell types vary across disease stages, further dictating differences in miRNA function. During the acute injury phase of AHF, cardiomyocytes and vascular endothelial cells serve as the primary targets of miRNA regulation. For example, miRNA-377, processed from pre-miR-377, inhibits angiogenesis by targeting vascular endothelial growth factor A (VEGFA) mRNA, playing a role in the acute vascular injury response[28]. During the long-term remodeling of CHF, the sustained activation of cardiac fibroblasts (CFs) becomes central. Thus, miRNAs that regulate fibroblast function are critical[27,29]. Meanwhile, infiltrating immune cells and their mediated inflammatory responses are also key targets in the chronic phase, with relevant miRNAs involved in their regulation[30]. Additionally, cells in distal organs such as hippocampal neurons and skeletal muscle cells are integrated into the miRNA regulatory network, forming complex systemic disease manifestations[31].
BIOLOGICAL CHARACTERISTICS AND MECHANISMS OF ACTION OF miRNAs
Biological synthesis of miRNAs
miRNA synthesis consists of two stages: primary transcription in the nucleus and maturation in the cytoplasm. In the nucleus, miRNA genes are transcribed into primary miRNAs (pri-miRNAs) with hairpin structures under the catalysis of RNA polymerase II. Subsequently, pri-miRNAs are cleaved by the RNase III family enzyme Drosha and its cofactor DiGeorge syndrome critical region gene 8 (DGCR8) into pre-miRNAs of 70-100 nucleotides in length. Pre-miRNAs are transported to the cytoplasm by the nuclear export protein Exportin-5 and further cleaved by another RNase III family enzyme Dicer into double-stranded miRNAs (miRNA/miRNA*) of 18-25 nucleotides. Among them, the complementary strand miRNA* is rapidly degraded. Finally, the mature single-stranded miRNA binds to Argonaute (AGO) proteins to form the RNA-induced silencing complex (RISC), which serves as the core structure for miRNA function[32-34].
Mechanisms of action of miRNAs
The core mechanism of miRNA action is the regulation of target gene expression at the post-transcriptional level. The regulatory mode mainly depends on the degree of base complementarity between the mature miRNA and the target gene mRNA, and also requires the mediation of the RISC. Specifically, it can be divided into two main ways. The first is the complete or near-complete complementarity mode, which is common in plant miRNAs but rare in animals. When the miRNA achieves complete complementarity with the coding region or 3'UTR of the target gene mRNA, it activates the endonuclease activity in RISC, directly cleaving the phosphodiester bond of the target mRNA, leading to the degradation of the target mRNA and thus rapidly blocking the gene expression process. The second is the incomplete complementarity mode, which is the most important regulatory way of miRNAs in animals. At this time, there is usually only partial base complementarity between the miRNA and the 3'UTR of the target gene mRNA. In particular, the complementarity between the "seed region" (nucleotides 2-8 at the 5' end of the miRNA) and the target mRNA is crucial. This incomplete pairing does not trigger the direct degradation of the target mRNA but inhibits gene expression through two pathways: first, after RISC binds to the target mRNA, it hinders the binding of ribosomes to the mRNA or interferes with the movement of ribosomes, thereby inhibiting the initiation and elongation of target gene translation; second, RISC can recruit related factors such as deadenylase to promote the degradation of the 3' polyadenylate tail of the target mRNA, accelerating the destabilization process of the target mRNA, making it more susceptible to recognition and degradation by intracellular nucleases, and indirectly reducing the expression level of the target gene[35-39].
In addition to classical intracellular regulation, miRNAs can also be released into the extracellular environment via extracellular vesicles (EVs) such as exosomes and microvesicles (MVs), or by forming complexes with high-density lipoprotein (HDL) and AGO proteins. These "circulating miRNAs" can stably persist in body fluids and may act as signaling molecules in intercellular communication, participating in remote organ crosstalk and the systemic regulation of pathological processes[40,41].
miRNAs IN ACUTE HEART FAILURE: ORCHESTRATORS OF DAMAGE ALERTS AND INFLAMMATORY STORMS
Acute myocardial injury, cardiomyocyte apoptosis and inflammatory storm together form a vicious cycle driving the rapid deterioration of cardiac function in the pathogenesis of AHF. Numerous miRNA-mediated mechanisms underlying AHF have been identified [Table 1].
miRNAs associated with AHF
| miRNAs | Experimental model | Effect | Mechanism of action | References |
| Myocardial cell apoptosis | ||||
| miR-143-3p | MI/R in mouse | Promoting | Inhibits Bcl-2 | [46] |
| miR-19b-3p | MI/R in mouse | Promoting | Inhibits PTEN | [47] |
| miR-489 | MI/R in mouse | Promoting | Inhibits SPIN1 | [48] |
| miR-100-5p | MI/R in mouse | Promoting | Inhibits PRMT5 | [49] |
| miR-133a/b | MI in rats; NRCMs + H2O2; H9c2 + H/R | Attenuating | Inhibits Caspase-9, Bax; upregulates Bcl-2 | [50-54] |
| miR-21 | MI in rats; H9c2 + H/R | Attenuating | Inhibits PDCD4 | [55-57] |
| miR-346 | MI/R in rats | Attenuating | Inhibits Bax | [58] |
| miR-499 | LPS in rats | Attenuating | Inhibits SOX6 and PDCD4 | [59] |
| miR-144 | MI/R in mouse; H9c2 + H/R | Attenuating | Inhibits TMEM16A Ca2+-activated chloride channel | [60] |
| miR-142-3p | MI/R in mouse; NRCMs + H2O2 | Attenuating | Inhibits expression and activation of TLR4/NF-κB pathway | [61] |
| miR-486-5p | CME in rats | Attenuating | Inhibits PTEN; activates PI3K/Akt signaling pathway | [62] |
| Inflammatory response | ||||
| miR-155 | AMI in mouse | Activating | Inhibits SOS1 | [63] |
| miR-4763-3p | AC16 + LPS | Activating | Inhibits IL10RA | [64] |
| miR-484 | H9c2 + LPS | Activating | Inhibits Yap1 | [65] |
| miR-126 | MI/R in rats; AC16 + oxygen-glucose deprivation/recovery (OGD/R) | Inhibiting | Activates GSK-3β signaling pathway | [66] |
| miR-221/-222 cluster | Coxsackievirus B3 (CVB3) in mouse | Inhibiting | ETS1/2; IRF2 TOX; CXCL12 | [67] |
| miR-146a | IL-1β in mouse | Inhibiting | Inhibits MAPK4/Drp-1 signaling pathway | [68] |
Acute myocardial injury and cardiomyocyte apoptosis
Acute myocardial injury is one of the common causes of AHF, often triggered by conditions such as AMI or viral myocarditis[42,43]. During this process, cardiomyocyte apoptosis serves as an important mechanism leading to the deterioration of cardiac function[44,45]. A subset of miRNAs is upregulated in myocardial ischemia/ reperfusion (MI/R) injury, acting as accelerators of cardiomyocyte apoptosis by inhibiting anti-apoptotic factors or activating pro-apoptotic signals. The mitochondrial pathway is central to cardiomyocyte apoptosis. miR-143-3p expression increases over time in myocardial tissue post-reperfusion. By directly targeting and inhibiting the anti-apoptotic protein B-cell Lymphoma 2 (Bcl-2), it disrupts mitochondrial membrane potential, promotes cytochrome c release, and activates caspase proteins, thereby exacerbating mitochondria-mediated apoptosis[46]. Similarly, miR-19b-3p downregulates phosphatase and tensin homolog (PTEN) gene expression, altering the balance between downstream apoptosis-related proteins Bax and Bcl-2 to promote cardiomyocyte apoptosis[47]. miR-489 facilitates apoptosis by targeting and inhibiting spindlin 1 (SP1N1), which in turn suppresses the downstream phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) survival signaling pathway[48]. Furthermore, miR-100-5p is significantly upregulated in ischemia/reperfusion (I/R)-injured myocardial tissue. By downregulating protein arginine methyltransferase 5 (PRMT5), it impairs PRMT5-mediated methylated inactivation of PTEN, leading to enhanced PTEN activity. This inhibits the PI3K-AKT survival pathway and ultimately exacerbates cardiomyocyte apoptosis[49].
Another subset of miRNAs exerts protective effects in acute cardiac injury. They act as cellular guardians by inhibiting pro-apoptotic signals or enhancing cell viability. miR-133 is a key endogenous inhibitor of cardiomyocyte apoptosis. Studies have confirmed that miR-133 can directly bind to and inhibit the expression of caspase-9, thereby blocking the downstream activation of caspase-3 in models of MI/R injury or oxidative stress[50]. Additionally, miR-133 regulates Bcl-2 family proteins by upregulating the anti-apoptotic protein Bcl-2 and downregulating the pro-apoptotic protein Bax. This inhibits the release of cytochrome C, thereby maintaining mitochondrial membrane stability and preventing the initiation of cell apoptosis[51]. In multiple models including MI/R injury, oxidative stress, hypoxia/reoxygenation (H/R), and isoflurane preconditioning, the upregulation of miR-21 significantly attenuates cardiomyocyte apoptosis by inhibiting programmed cell death 4 (PDCD4) expression[52-54]. Furthermore, miR-21 can regulate the FAS-mediated apoptotic pathway by inhibiting homeodomain-interacting protein kinase 3 (HIPK3)[55], and activate the pro-survival PI3K/Akt signaling pathway by suppressing PTEN, forming its multi-layered anti-apoptotic network[56,57]. However, in the fibrotic progression of CHF (Section "Myocardial fibrosis"), the persistently overexpressed miR-21 exerts a profibrotic and detrimental effect by acting on fibroblasts and regulating the transforming growth factor-beta (TGF-β)/small mother against decapentaplegic (SMAD) signaling pathway, which highlights its highly contextdependent function. Studies have also found that miR-346 expression is downregulated in MI/R injury models, while its overexpression attenuates cardiomyocyte apoptosis and reduces infarct size by targeting and inhibiting the key pro-apoptotic protein Bax[58]. In lipopolysaccharide (LPS)-induced models of septic cardiomyopathy, miR-499 expression is suppressed, leading to the upregulation of its target genes SRY-box transcription factor 6 (SOX6) and PDCD4 and thereby promoting cardiomyocyte apoptosis[59]. In MI/R models, miR-144 exerts acute cardioprotective effects by inhibiting the transmembrane protein 16A (TMEM16A) Ca2+-activated chloride channel to reduce myocardial infarction size[60]. miR-142-3p expression is downregulated in hearts with MI/R injury, and its overexpression inhibits the expression and activation of the toll-like receptor 4 (TLR4)/nuclear factor-κB (NF-κB) pathway, significantly reducing cardiomyocyte apoptosis rate and improving cardiac function[61]. Additionally, miR-486-5p activates the PI3K/Akt signaling pathway by targeting and inhibiting the PTEN gene, thereby suppressing cardiomyocyte apoptosis induced by coronary microembolism (CME) in rats[62].
Inflammatory response
Following acute myocardial injury, damaged cells release damage-associated molecular patterns (DAMPs), thereby triggering robust activation of the innate immune system. This is characterized by the rapid infiltration of neutrophils and monocytes/macrophages, as well as the explosive release of pro-inflammatory cytokines such as interleukin (IL)-1β and tumor necrosis factor-α (TNF-α). Moderate inflammation facilitates the clearance of necrotic tissue; however, excessive inflammatory responses exacerbate myocardial injury, expand the infarct size, and initiate adverse ventricular remodeling. miRNAs serve as key regulatory hubs in this pathophysiological process.
Some miRNAs can enhance inflammatory responses and aggravate myocardial injury by regulating related pathways. In an AMI model, the expression of the miR-155 precursor is upregulated in macrophages, which then secrete exosomes enriched with miR-155. These exosomes downregulate son of sevenless homolog 1 (SOS1) protein expression and reduce suppressors of cytokine signaling 1 (SOCS1) levels, thereby promoting inflammation while inhibiting the proliferation of CFs, ultimately exacerbating myocardial injury[63]. In a LPS-induced cardiomyocyte inflammation model, miR-4763-3p expression is elevated. By targeting and suppressing interleukin-10 receptor A (IL10RA), it leads to increased levels of pro-inflammatory factors IL-6 and IL-1β, along with decreased levels of anti-inflammatory factors IL-10 and TGF-β, thereby aggravating the inflammatory response[64]. Furthermore, in LPS-treated H9c2 (2-1) rat embryonic cardiomyocyte cell line (H9C2) cells, miR-484 expression is upregulated. It directly targets the mRNA of Yes-associated protein 1 (Yap1), inhibiting cell viability, promoting apoptosis, and activating the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, resulting in increased secretion of inflammatory factors such as TNF-α, IL-1β, and IL-6[65].
Other miRNAs exert cardioprotective effects through anti-inflammatory mechanisms, thereby delaying the progression of AHF. For instance, Guo et al. demonstrated in a rat model of MI/R injury that transfection of miR-126 reduces the production of inflammatory factors such as IL-6, IL-8, and TNF-α, and attenuates myocardial injury and inflammatory responses by modulating the glycogen synthase kinase-3β (GSK-3β) signaling pathway[66]. In viral myocarditis, the miR-221/-222 cluster is upregulated in cardiomyocytes, playing a dual role in antiviral defense and inflammation regulation. This cluster targets multiple genes including E26 transformation-specific transcription factor 1/2 (ETS1/2), interferon regulatory factor 2 (IRF2), thymocyte selection-associated high mobility group box (TOX), and C-X-C motif chemokine ligand 12 (CXCL12), thereby suppressing viral replication and modulating inflammatory responses. Inhibition of this cluster leads to increased viral load, exacerbated inflammation, and aggravated cardiac injury[67]. Furthermore, miR-146a levels are elevated in exosomes derived from IL-1β-stimulated macrophages. These exosomes alleviate myocardial damage and improve cardiac function in septic mice by regulating mitochondrial homeostasis, enhancing oxidative stress resistance, and exerting anti-inflammatory effects[68].
miRNAs IN CHRONIC HEART FAILURE: PERSISTENT DRIVERS OF ADVERSE VENTRICULAR REMODELING
Myocardial remodeling constitutes the core pathophysiological alteration in CHF, which is specifically manifested by cardiomyocyte hypertrophy, myocardial fibrosis, ventricular chamber dilatation, and a progressive decline in cardiac contractile function. Several miRNAs are modulated and participate in the regulation of CHF [Table 2].
miRNAs associated with CHF
| miRNAs | Experimental model | Effect | Mechanism of action | References |
| Cardiac hypertrophy | ||||
| miR-1 | NRCMs + T3 or phenylephrine | Inhibition | Suppress HDAC4; inhibits twinfilin-1 | [69,70] |
| miR-133 | NRCMs+ thyroid hormone | Inhibition | Inhibits SERCA2a and calcineurin | [71] |
| miR-222 | TAC in mouse | Inhibition | inhibits PUMA, HMBOX1 and NFATc3 | [72] |
| miR-31-5p | Abdominal aorta coarctation in rats | Inhibition | Inhibits the co-localization of Nfatc2ip and β-Mhc | [73] |
| miR-30d | Isoproterenol in mouse; NRCMs + phenylephrine or Ang II | Inhibition | Inhibits MAP4K4, GRP78 and hypertrophic nuclear factor NFAT | [74] |
| miR-208a | TAC in mouse | Promotion | Inhibits antihypertrophic GATA4 | [75] |
| miR-155 | TAC in mouse | Promotion | Inhibits SOCS1 | [76] |
| miR-34c-5p | Isoprenaline in mouse; NRCMs + Isoprenaline | Promotion | Inhibits autophagy-related gene ATG4B | [77] |
| miR-495-3p | Aortic banding in rats; H9c2 + Ang II | Promotion | Inhibits Pum2 | [78] |
| Cardiac fibrosis | ||||
| miR-133 family | Streptozotocin in mouse | Inhibition | Suppresses COL1A1; inhibits CTGF | [80,81] |
| miR-29 family | MI in mouse | Inhibition | Inhibits TGFβ, VEGFA and DNMT3A | [82,83] |
| miR-146a-5p | MI in mouse | Inhibition | Inhibits TXNIP and EndMT | [84] |
| miR-17-5p | Abdominal aorta coarctation in mouse; CFs + Ang II | Inhibition | Inhibits BNIP3 | [85] |
| miR-486a-5p | CFs + IgE | Inhibition | Inhibits Smad1 pathway | [86] |
| miR-21 | MI in mouse; CFs + TGF-β1 | Promotion | Activates AKT pathway; TGF-β/SMAD pathway | [87,88] |
| miR-208a | MI in rats | Promotion | DYRK2 | [89] |
| miR-15a-5p | Ang II in mouse; CFs + Ang II | Promotion | Dephosphorylated NFAT by targeting DYRK2 | [90] |
Myocardial hypertrophy
Cardiomyocyte hypertrophy is a key early pathological change in CHF, representing an adaptive response of myocardial tissue to long-term increased pressure or volume load. However, persistent pathological hypertrophy will eventually lead to cardiomyocyte dysfunction and even apoptosis, further exacerbating the deterioration of cardiac function. By regulating the expression of target genes in signaling pathways related to myocardial cell hypertrophy, miRNAs play an important regulatory role in the process of myocardial hypertrophy.
The miRNA pair miR-133 and miR-1, which are transcribed from the same genetic unit, are consistently downregulated in hypertrophic hearts. Overexpression of miR-1 can effectively suppress cardiomyocyte hypertrophy induced by stimuli such as thyroid hormone (T3)[69] and norepinephrine[70]. In mouse models of thyroid hormone-induced cardiac hypertrophy, miR-133 mimics markedly inhibit cardiomyocyte hypertrophy via its target genes sarcoplasmic/endoplasmic reticulum calcium ATPase 2a (SERCA2a) and calcineurin[71]. miR-222 is markedly upregulated in transverse aortic constriction (TAC)-induced models of pathological myocardial hypertrophy and heart failure. Its overexpression mitigates pressure overload-induced pathological hypertrophy, improves cardiac function and boosts survival rates, while its inhibition accelerates heart failure progression. These effects are mediated by targeting the pro-apoptotic factor p53 upregulated modulator of apoptosis (PUMA) and the transcription factors homeobox containing 1 (Hmbox1) and nuclear factor of activated T cells cytoplasmic 3 (NFATc3)[72]. miR-31-5p was found to exert an anti-hypertrophic effect in a rat model of cardiac hypertrophy induced by abdominal aortic constriction, an effect involving nuclear factor of activated T cells 2 interacting protein (NFATC2IP), which binds to and enhances the transcriptional activity of the hypertrophy marker gene β-myosin heavy chain (β-Mhc)[73]. miR-30d expression is reduced both in hypertrophy models and in the serum of patients with chronic heart failure. Overexpression of miR-30d ameliorates cardiac hypertrophy by targeting mitogen-activated protein kinase kinase kinase 4 (MAP4K4) and glucose-regulated protein 78 (GRP78), and by inhibiting the nuclear factor of activated T cells (NFAT) signaling pathway[74].
Some miRNAs are upregulated under pathological stimuli such as pressure overload and neurohormonal activation, directly promoting cardiomyocyte hypertrophy and adverse remodeling. As a cardiac-specific miRNA, miR-208a is markedly upregulated in response to hemodynamic stress. In rodent models, both genetic ablation and pharmacological inhibition of miR-208a have been demonstrated to attenuate stress-induced cardiac hypertrophy and remodeling; conversely, cardiac-specific transgenic overexpression of miR-208a alone is sufficient to induce cardiomyocyte hypertrophy[75]. Macrophage-derived miR-155 promotes cardiomyocyte hypertrophy and dysfunction in a paracrine manner by targeting and suppressing suppressor of SOCS1. Inhibition of miR-155 in macrophages significantly attenuates pressure overload-induced cardiac inflammation, hypertrophy, and dysfunction[76]. miR-34c-5p is upregulated in an isoproterenol-induced cardiac hypertrophy model. It promotes hypertrophy development by directly targeting autophagy-related gene 4B (ATG4B) and inhibiting autophagic flux in cardiomyocytes[77]. miR-495-3p is highly expressed in an aortic constriction rat model and in angiotensin II (Ang II)-stimulated cardiomyocytes. Silencing miR-495-3p alleviates hypertrophy by targeting Pumilio RNA-binding family member 2 (Pum2)[78].
Myocardial fibrosis
Myocardial fibrosis is another core pathological change of CHF, characterized by excessive activation and proliferation of myocardial fibroblasts and secretion of a large amount of extracellular matrix proteins, ultimately leading to cardiac structural remodeling and functional failure[79]. Several miRNAs are regulated and involved in the regulation of myocardial fibrosis.
Among the miRNAs that negatively regulate myocardial fibrosis, the miR-133 and miR-29 families play a core regulatory role. In diabetic cardiomyopathy, miR-133a expression is reduced. Treatment with human umbilical cord mesenchymal stromal cells improves myocardial fibrosis by restoring the expression of miR133a and its target mRNA collagen type I alpha 1 chain (COL1A1)[80]. While, miR133b ameliorates fibrosis by targeting connective tissue growth factor (CTGF), thereby inhibiting the activation and proliferation of CFs[81]. The miR-29 family serves as a potent antifibrotic factor. miR29a exerts its inhibitory effect by suppressing the expression of TGFβ and VEGFA[82]. Studies have developed an exosomeloaded microneedle patch carrying miR29b mimics for localized delivery after myocardial infarction, which effectively reduces fibrosis and improves cardiac function[83]. miR-146a-5p is downregulated in human cardiac microvascular endothelial cells (HCMECs) under hypoxic conditions. However, its overexpression can inhibit cardiac fibrosis after myocardial infarction by targeting thioredoxin-interacting protein (TXNIP) and suppressing endothelial-to-mesenchymal transition (EndMT)[84]. In a mouse model of myocardial fibrosis induced by abdominal aortic constriction, miR-17-5p directly targets the 3'-UTR of BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), significantly downregulating its expression, restoring mitochondrial balance, and thereby alleviating pathological cardiac fibrosis[85]. Additionally, overexpression of miR-486a-5p can mitigate Ang II-induced myocardial fibrosis by targeting Smad1[86].
miR-21 is one of the pro-fibrotic miRNAs that have been widely studied. In rat models of myocardial infarction (MI) and TGF-β1-treated cells, miR-21-containing MVs are overexpressed and promote cardiac fibrosis by activating the AKT pathway[87]. Additionally, miR-21 can also promote myocardial fibrosis after MI by targeting the TGF-β/SMAD family member 7 (Smad7) signaling pathway[88]. In a rat model of myocardial infarction, inhibition of miR-208a via adeno-associated virus serotype 9 (AAV9) markedly attenuates cardiac fibrosis and improves cardiac function[89]. miR-15a-5p is upregulated in cardiomyocytes stimulated by Ang II. Through exosomal transport to CFs, it targets dual specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2) to dephosphorylate NFAT, thereby promoting fibroblast activation and the expression of collagen genes COL1A1 and collagen type III alpha 1 chain (COL3A1)[90].
Commonalities and differences of miRNAs in acute and chronic heart failure
The roles of miRNAs in HF exhibit stagespecific duality: the same miRNA can exert identical or opposing effects in acute versus chronic disease. This duality arises from stage-specific physiological demands, target-cell shifts, and microenvironmental[91] changes. Here, we analyze two paradigmatic miRNAs, miR-133 and miR-21, to systematically dissect how their dual roles are governed by disease stage and cellular context [Table 3].
Comparison of key miRNAs in acute and chronic heart failure
| miRNAs | Expression/function in AHF | Expression/function in CHF | Key targets/pathways |
| miR-133 | Downregulated; anti-apoptotic | Downregulated; anti-hypertrophic and anti-fibrotic | Caspase-9, Bax/Bcl-2; SERCA2a, calcineurin |
| miR-21 | Upregulated; anti-apoptotic | Upregulated; pro-fibrotic | PDCD4, PTEN/PI3K/AKT; TGF-β/SMAD |
| miR-155 | Upregulated; pro-inflammatory | Upregulated; promotes hypertrophy | SOS1/SOCS1 |
miR-133: from acute anti-apoptosis to chronic anti-remodeling
miR-133 shows the core commonality of protective effects in AHF and CHF. The core pathological essence of heart failure resides in the structural damage and functional loss of cardiomyocytes, followed by abnormal global myocardial remodeling[92], and miR-133 directly targets this core pathological process in both AHF and CHF. In the AHF phase, cardiomyocyte apoptosis is the pivotal driver of acute myocardial injury and rapid cardiac dysfunction[93]. miR-133 directly mitigates the acute loss of cardiomyocytes by inhibiting apoptosis, thereby preserving cardiomyocyte survival and basic biological functions. However, during the progression of the disease, excessive myocardial hypertrophy and fibrosis can become important driving factors for the deterioration of HF[91]. miR-133 maintains the normal morphology and function of cardiomyocytes and safeguards the overall structural integrity of myocardial tissue through suppressing hypertrophy and fibrosis.
Furthermore, AHF and CHF are not discrete pathological stages but a continuous progression in the development of HF. If cardiomyocyte apoptosis is not effectively inhibited in AHF, extensive loss of cardiomyocytes will trigger compensatory myocardial hypertrophy, which further progresses to myocardial hypertrophy and fibrosis in CHF and ultimately leads to progressive cardiac dysfunction[94]. miR-133 blocks acute cardiomyocyte apoptosis in the AHF phase, thus suppressing the initiation and rapid deterioration of heart failure at the upstream level; in the CHF phase, it abrogates myocardial hypertrophy and fibrosis, curbing the chronic progression of heart failure at the downstream level. In both phases, miR-133 exerts a global antagonistic effect on heart failure by targeting key links in the heart failure pathological cascade, rather than merely intervening in a single pathological stage.
miR-21: from acute cardioprotection to chronic pro-fibrosis
miR-21 exhibits a dual role in HF, with conflicting conclusions across different studies, which fundamentally stems from its stagedependent functional dichotomy. Most preclinical models, by targeting CFs, have confirmed its profibrotic role in CHF[95,96]. However, Wang et al. reported that miR-21 plays a protective role in murine AMI[97]. This discrepancy arises from two key factors. First, the disease subtypes investigated differ. The study by Wang et al. focused on ischemic heart failure, where the core driver of dysfunction remains residual cardiomyocyte apoptosis, whereas pressureoverload models primarily simulate the chronic remodeling process centered on fibrosis[97]. Second, the cellular targets of miR-21 undergo dynamic shifts with disease progression. In the AHF stage, upregulated miR-21 is predominantly localized to cardiomyocytes, where its function centers on autonomous protection, with low expression in CFs. Upon progression to the CHF stage, miR-21 expression significantly shifts towards CFs. By directly targeting Smad7 mRNA, it relieves the inhibition on the TGFβ signaling pathway, thereby amplifying fibrotic remodeling. Furthermore, miR-21 can promote pathological cardiomyocyte hypertrophy by activating the extracellular regulated protein kinase/mitogen-activated protein kinase signaling pathway[98]. In the context of CHF that has progressed to the decompensated phase, this prohypertrophic effect further accelerates the vicious cycle of hypertrophyfibrosisworsening cardiac function. These seemingly contradictory findings collectively reveal that the function of miR-21 is highly dependent on the specific disease stage and cellular microenvironment. Therefore, in developing miRNAbased therapies, it is essential to perform meticulous stratification based on the subtype and stage of heart failure to enable tailored interventions.
miRNA REGULATORY NETWORK IN HEART FAILURE
The "cell survival/death decision-making network" in AHF
At the core of acute myocardial injury lies a molecular decision-making network governed by the dynamic equilibrium between pro-apoptotic and anti-apoptotic miRNAs. For instance, during MI/R injury, pro-apoptotic miRNAs, miR-143-3p and miR-19b-3p, attenuate cell survival signals by inhibiting targets such as Bcl-2 or PTEN. Concurrently, anti-apoptotic miRNAs, miR-133, miR-21, and miR-499 are rapidly induced or basally expressed, activating survival pathways such as PI3K/Akt through the inhibition of targets including Caspase-9, PDCD4, or SOX6. These miRNAs do not act in isolation; rather, their actions converge at the critical crossroads of the mitochondrial apoptotic pathway and the PI3K/Akt pathway (Figure 1, left panel). Both miR-19b-3p and miR-21 target PTEN. However, upregulation of the former promotes apoptosis in the acute phase, while upregulation of the latter enhances survival signaling. This suggests that their functions are likely regulated by more precise temporal or spatial cues. Understanding the disruption and restoration of this network balance is crucial for developing protective intervention strategies during the acute phase.
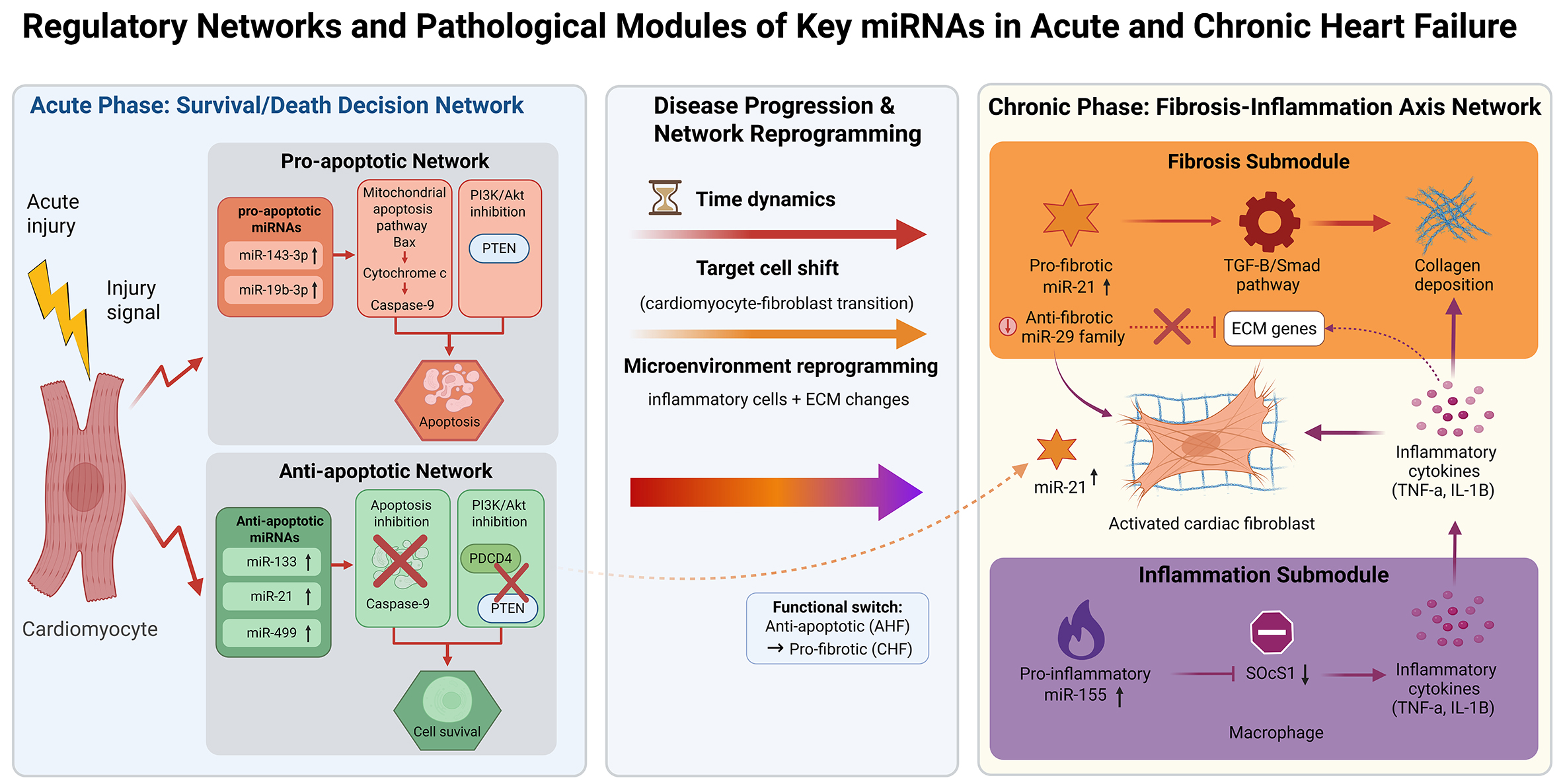
Figure 1. Regulatory networks and pathological modules of key miRNAs in acute and chronic heart failure. Created with BioRender.com (Created in BioRender. Nû, _à. (2026)) (https://www.biorender.com/lh4iftz).
The "fibrosis-inflammation axis network" in CHF
The hallmark of chronic ventricular remodeling is sustained low-grade inflammation and progressive fibrosis, processes tightly coordinated by an interacting network of miRNAs. The pro-fibrotic hub miR-21 is highly expressed in CFs, where it directly drives collagen deposition by inhibiting Smad7, thereby releasing the brake on the TGF-β/Smad pathway. Simultaneously, expression of the anti-fibrotic miR-29 family is suppressed, which relieves its potent repression of numerous extracellular matrix protein genes, functionally antagonizing the action of miR-21. Furthermore, the pro-inflammatory miR-155 sustains the inflammatory phenotype of macrophages by targeting suppressors such as SOCS1. The resulting inflammatory cytokines can further activate CFs and upregulate miR-21, thereby linking the inflammatory and fibrotic networks into a self-amplifying vicious cycle (Figure 1, right panel). Targeting multiple nodes within this network, the combined inhibition of miR-21 and the restoration of miR-29 expression may be more effective in preventing the progression of fibrosis than single-target intervention.
This schematic summarizes the key miRNA regulatory networks central to the pathological modules of AHF and CHF. In the AHF phase (left panel), a balance is formed within cardiomyocytes between pro-apoptotic and anti-apoptotic miRNAs, which jointly regulate the mitochondrial apoptosis and PI3K/Akt survival pathways to determine cell fate. In the CHF phase (right panel), pro-fibrotic miRNAs (miR-21), anti-fibrotic miRNAs (the miR-29 family), and pro-inflammatory miRNAs (miR-155) constitute an interconnected fibrosis-inflammation axis network, driving adverse ventricular remodeling. The central panel highlights how changes in time, target cells, and microenvironment during disease progression from acute to chronic stages lead to a reprogramming of the entire regulatory network, with the functional shift of miR-21 serving as a representative example. Arrows indicate activation, promotion, or directionality; flat-ended lines denote inhibition. ECM: Extracellular matrix. Created in BioRender (BioRender.com).
CLINICAL APPLICATION PROSPECTS OF miRNAs IN HEART FAILURE
As a diagnostic and prognostic biomarker
Although traditional biomarkers of heart failure such as B-type natriuretic peptide (BNP) and N-terminal pro-B-type natriuretic peptide (NT-proBNP) are sensitive, they have insufficient specificity and limitations in prediction or early diagnosis[99]. Circulating miRNAs have become highly promising new biomarkers due to their stability, tissue specificity and close association with pathological processes. Individual or combined miRNA panels can effectively distinguish between HF patients and healthy individuals, and further discriminate among different HF subtypes. Several studies have shown that miR-19b, miR-21, and miR-208a exhibit significant expression changes in the plasma of HF patients, demonstrating high sensitivity and specificity in distinguishing between heart failure with mildly reduced ejection fraction (HFmrEF) and HFpEF[100]. A large-scale clinical study developed an 8-miRNA combined panel, which, when used in conjunction with NT-proBNP, significantly improves the specificity and accuracy of diagnosing non-acute HF. It can even reclassify cases that might be missed based solely on NT-proBNP thresholds[101].
Specific miRNA profiles correlate with different stages and subtypes of HF. For example, miR-30d expression is associated with left ventricular systolic and diastolic volumes, miR-126-3p with pulmonary artery pressure, and miR-483-3p with right ventricular remodeling and central venous pressure[102]. This suggests that different miRNAs may reflect specific cardiac structural or hemodynamic abnormalities during HF progression, facilitating more refined disease stratification and early warning.
As therapeutic targets
Based on the key regulatory role of miRNAs in the occurrence and development of HF, regulating miRNA expression has become a novel strategy for HF treatment. Currently, therapeutic approaches targeting miRNAs mainly include miRNA mimics and miRNA inhibitors. The core miRNA classification and their clinical translation potential can be found in Table 4.
miRNA subclassification based on pathological mechanisms and transformation potential
| Representative miRNA | Classification | Effects | Treatment strategy |
| miR-133 & miR-499 | Acute cardioprotection | Anti-apoptosis | miRNA mimics |
| miR-155 | Inflammatory regulation | Pro-inflammation | antimiR |
| miR-30d | sophisticated delivery technology | Anti-hypertrophy and fibrosis | Engineered exosome delivery (EVs; Nanoparticles; Liposome) |
miRNA mimics
miRNA mimics are artificially synthesized oligonucleotides with the same sequence as endogenous mature miRNAs. They can mimic the biological functions of endogenous miRNAs and exert effects by targeting and regulating the expression of downstream genes. For miRNAs that are downregulated in HF, restoring their physiological expression levels through the infusion of miRNA mimics can effectively inhibit myocardial remodeling and protect cardiac function. For example, studies have confirmed that intravenous injection of miR-30d-modified milk-derived exosomes (miR30d-mEVs-IMTP) can target and deliver miR-30d to diseased cardiac tissue, significantly alleviating myocardial hypertrophy, inflammatory responses, and fibrosis during HF progression[103].
miRNA inhibitors
For miRNAs abnormally upregulated in HF, inhibition using antisense oligonucleotides (antimiRs) represents the primary therapeutic strategy. Among these approaches, therapies targeting miR-132 stand out as the most compelling. Preclinical studies have confirmed that miR-132 is a key driver of pathological myocardial hypertrophy and adverse cardiac remodeling[104]. A first-in-human Phase Ib randomized controlled trial demonstrated that the antisense oligonucleotide drug CDR132L safely and effectively reduces plasma miR-132 levels in patients with chronic ischemic heart failure. This reduction was accompanied by a decrease in NT-proBNP, narrowing of the QRS complex (QRS), and a trend toward improved cardiac fibrosis biomarkers[105,106]. This milestone marks a pivotal step in translating miRNA-based HF therapies from conceptual framework to clinical practice.
Core barriers to clinical translation and emerging solutions
Despite the promising regulatory role of miRNAs in HF, their clinical translation is hindered by several critical technical barriers, particularly in delivery efficiency and safety control. Addressing these barriers is essential for advancing miRNA-based therapies from preclinical research to clinical application.
Endosomal escape and targeting specificity
A major bottleneck in miRNA-based therapy lies in the poor in vivo stability and inefficient delivery of naked miRNA mimics or inhibitors. These unmodified oligonucleotides are highly susceptible to nuclease degradation and fail to penetrate target cells effectively, limiting their biological activity[107]. Even when delivered via carriers such as liposomes or polymers, most miRNAs are trapped in the endosome-lysosome pathway and degraded, preventing their translocation to the cytoplasm where they exert regulatory functions[107]. Additionally, systemic administration often leads to non-specific distribution in organs such as the liver and spleen rather than the diseased tissues, reducing therapeutic efficacy and increasing the risk of off-target toxicity[108]. For cardiovascular diseases involving deep tissue targets, the lack of tissue-specific homing further exacerbates delivery inefficiency[109].
To address these critical limitations, the rapid development of emerging delivery technologies aims to enhance stability, improve targeting, and facilitate kernel body escape. Biomimetic nanoparticles leverage cell membrane coating to inherit the biological properties of source cells, such as immune evasion and inherent tissue homing capacity. For AMI-induced AHF, macrophage membrane-coated nanoparticles combined with ultrasound-targeted microbubble destruction (UTMD) technology have been successfully used to deliver mesenchymal stem cell membrane proteins and miR-125b to inflamed injured regions of AMI. Notably, this strategy exhibited no significant toxicity in mouse models, underscoring its safety profile for acute cardiac injury intervention[110].
EVs are naturally secreted membranous vesicles with low immunogenicity, excellent biocompatibility, and intrinsic targeting abilities, making them highly promising delivery tools for HF therapy[107,111]. Specifically, miR-125a-5p encapsulated in mesenchymal stem cell-derived exosomes (MSC-Exos) has been validated to exert cardioprotective effects in MI/R injury by mitigating cardiomyocyte damage and regulating inflammatory responses[112]. Furthermore, active targeting to pathological cardiac tissues is achieved by conjugating delivery carriers with cell-specific antibodies, ligands, or aptamers. For example, cardiac-targeting peptide (CTP)-modified mesoporous silica (MSN) nanospheres loaded with miR-199a-5p can specifically home to ischemic myocardial tissue. This targeted delivery significantly inhibits cardiomyocyte apoptosis, enhances myocardial contractility, reduces infarct size post-myocardial infarction, and improves cardiac function[113].
The mechanism of miRNA action inherently confers a risk of off-target effects. A single miRNA typically targets multiple mRNAs, and one mRNA may be regulated by multiple miRNAs[111]. Exogenously introduced miRNA mimics or inhibitors, which function similarly to endogenous miRNAs, may bind to the 3' UTR of unintended targets via their seed sequence, leading to unexpected gene silencing or activation and inducing toxicity[114]. This highlights the potential off-target toxicity of RNA interference-based therapies. Specificity can be enhanced and off-target effects mitigated through the following strategies. Chemical modifications of miRNA oligonucleotides, such as locked nucleic acid (LNA), 2'-O-methyl, or phosphorothioate modifications, can significantly improve nuclease stability, enhance binding affinity to target mRNAs, and sometimes reduce immunogenicity[115]. For instance, an LNA-modified antagomir (AM106) targeting miR-106b-5p effectively prevented doxorubicin-induced cardiotoxicity without compromising its antitumor efficacy in preclinical studies[115]. In addition, site-specific delivery via local injection[112], inhalation, or responsive materials[116] enables targeted release at pathological sites, reducing systemic exposure and off-target effects.
Immunogenicity
Immunogenicity poses a major safety barrier to miRNA therapy: certain viral vectors or synthetic nanoparticles may be recognized by the immune system, triggering inflammatory responses or the production of neutralizing antibodies, which impairs the efficacy of repeated administration[107]. The most effective mitigation strategy is the use of immunologically inert carriers that avoid immune recognition. EVs are nanoscale membrane-structured vesicles naturally secreted by cells. Their surface components are similar to those of the source cells and are rich in biomolecules such as phospholipids, proteins and sugars. This natural biocompatibility makes it less likely to be recognized as a foreign substance by the immune system in the body, thereby reducing the risk of an immune response. Hydrogels loaded with miR-222-engineered EVs can alleviate acute I/R injury and inhibit cardiac remodeling after I/R through mechanical conduction[117].
Past miRNA therapeutic failures and lessons for clinical translation
Liposomal miR-34a mimic (MRX34), the first miRNA-based cancer therapy to enter clinical trials, has provided invaluable experience and profound lessons for the entire research field. As a pioneering miRNA therapy for solid tumors, MRX34 is a miR-34a mimic delivered via lipid nanoparticles (LNPs) that advanced to Phase II clinical trials, yet it was prematurely terminated due to severe immune-related adverse events (IRAEs) and hepatotoxicity[118,119]. This case offers critical warnings and precious insights for the development of miRNA therapies for heart failure, particularly with respect to delivery strategies.
Although MRX34 was developed for cancer treatment, its target, miR-34a, plays a central role in the pathogenesis and progression of HF, and its delivery platform is also a commonly used vector for non-coding RNA therapeutics. The failure of MRX34 stems in part from an inadequate understanding of the extensive and complex functional network of miR-34a under normal physiological conditions and various pathological states. miR-34a is a key regulatory node in HF. Numerous studies have demonstrated that miR-34a is upregulated in various HF models and drives disease progression by repressing multiple key protective targets[120,121]. Thus, prior to developing miRNA therapies for heart failure, it is imperative to conduct systems biology studies that go beyond single-target analysis and comprehensively map the regulatory networks of the target miRNA across distinct cardiac cell types, different disease stages and various etiological factors of HF[121]. Blind repression of a miRNA that modulates multiple protective signaling pathways may disrupt normal physiological feedback mechanisms and induce unforeseen adverse effects.
Furthermore, MRX34 was administered via systemic LNP delivery, which lacks tissue specificity and constitutes the primary cause of its severe immunotoxicity. This represents an insurmountable barrier that must be overcome for the treatment of CHF. Therefore, investment is warranted in the development of next-generation delivery platforms with high cardiac tropism. This may involve the use of nanoparticles modified with cardiotropic peptides, antibodies or aptamers, or the application of smart materials that respond to the pathological microenvironment of the heart. Additionally, more sensitive preclinical models need to be established to assess off-target effects and immunogenicity of miRNA therapeutics.
LIMITATIONS AND CHALLENGES
Despite considerable progress in elucidating the roles of miRNAs in HF, several persistent limitations and translational challenges must be acknowledged and overcome to advance their clinical application. First, the functional effects of miRNAs are highly context-dependent, varying substantially across cell types, disease stages, and etiological backgrounds. Many current insights derive from animal models or in vitro systems, which may not fully mirror the pathophysiological complexity of human HF. Moreover, miRNA-target interactions are frequently predicted bioinformatically and lack rigorous experimental validation in relevant in vivo settings. Second, although circulating miRNAs hold promise as diagnostic and prognostic biomarkers, their clinical utility is constrained by methodological inconsistencies in sample collection, processing, and quantification. The absence of standardized protocols and universally accepted reference miRNAs undermines the reproducibility and cross-study comparability of reported findings. Third, the development of miRNA-based therapeutics faces multifold hurdles. Key issues include inefficient and nonspecific delivery to cardiac tissue, poor oligonucleotide stability, potential immunogenicity, off-target effects due to pleiotropic miRNA regulation, and a scarcity of robust clinical trial data. Finally, HF is a heterogeneous syndrome encompassing diverse etiologies and phenotypic presentations. Existing miRNA studies often generalize results across HF subtypes, thereby overlooking distinct miRNA signatures and regulatory mechanisms specific to different clinical categories. Future investigations should therefore adopt more stratified, subtype-aware research designs to improve pathological resolution and therapeutic precision.
CONCLUSION AND FUTURE DIRECTIONS
This review systematically elucidates recent advances in miRNA research in acute and chronic heart failure (AHF/CHF). It covers the posttranscriptional regulatory roles of miRNAs in core pathophysiological processes such as myocardial apoptosis, inflammation, hypertrophy, and fibrosis, as well as their
Despite significant progress achieved in basic and clinical research, translating miRNA-based findings into clinical practice remains hindered by unresolved challenges, including incomplete mapping of miRNA regulatory networks, insufficient diagnostic specificity of single miRNA markers, and technical barriers in targeted delivery systems, immunogenicity control, and off-target effect mitigation. To address these gaps and promote the sustainable development of the field, future studies should focus on the following key aspects based on the miRNA classification framework of pathological mechanisms and transformation potential, clarifying the stage-specific and cell-type-specific regulatory roles of miRNAs in HF, improving the accuracy and specificity of miRNA-based biomarkers through multi-omics integration, optimizing myocardial-specific targeted delivery systems to reduce immunogenicity and off-target effects, and strengthening large-scale preclinical verification and clinical trials to ensure the safety and efficacy of miRNA therapies. Meanwhile, it is necessary to learn lessons from past clinical failures to avoid repeated risks.
In summary, miRNAs are key regulators of AHF and CHF, and their in-depth study is of great significance for clarifying the pathological essence of HF and promoting the development of precise diagnosis and treatment strategies. With the continuous improvement of research technologies and the breakthrough of key technical bottlenecks, it is expected that miRNAs will revolutionize the diagnosis, treatment, and prognosis evaluation of HF, promote the realization of personalized HF management, and ultimately improve the clinical outcomes of patients with HF.
DECLARATIONS
Acknowledgments
The graphical abstract and Figure 1 was created with BioRender.com (Created in BioRender. Nû, à. (2026)) (https://BioRender.com/a176xxx).
Authors' contributions
Conceived the idea and designed the outline of the review: Ren Z, Cai B
Wrote and revised the initial draft of the manuscript: Ren Z,
Performed the literature search and data analysis: Ma W, Wang X, Liu Y, Gao X
Supervised the project and provided critical revisions: Cai B
All authors read and approved the final version of the manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This study was supported by the Heilongjiang Province Natural Science Foundation (ZL2024H017).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Špinar J, Špinarová L, Vítovec J. Pathophysiology, causes and epidemiology of chronic heart failure. Vnitr Lek. 2018;64:834-8.
2. Bozkurt B, Coats AJ, Tsutsui H, et al. Universal definition and classification of heart failure: a report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur J Heart Fail. 2021;23:352-80.
3. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. Circulation. 2022;145:18.
5. Hummel A, Empen K, Dörr M, Felix SB. De novo acute heart failure and acutely decompensated chronic heart failure. Dtsch Arztebl Int. 2015;112:298-310.
6. Martens P, Dupont M, Verbrugge FH, et al. Urinary sodium profiling in chronic heart failure to detect development of acute decompensated heart failure. JACC Heart Fail. 2019;7:404-14.
7. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843-54.
8. Pasquinelli AE, Reinhart BJ, Slack F, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86-9.
9. Reinhart BJ, Slack FJ, Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901-6.
10. Lewis BP, Shih I, Jones-rhoades MW, Bartel DP, Burge CB. Prediction of mammalian MicroRNA targets. Cell. 2003;115:787-98.
11. Ren Z, Liu Y, Cai A, et al. Cannabidiol represses miR-143 to promote cardiomyocyte proliferation and heart regeneration after myocardial infarction. Eur J Pharmacol. 2024;963:176245.
12. Yaping X, Guotian Y, Dandan J, Jintao D, Xinyi L, Zhikun G. Fibroblast-derived exosomal miRNA-133 promotes cardiomyocyte-like differentiation. Acta Histochem. 2022;124:151931.
13. Lee T, Shen W, Chen Y, et al. Mir221- and Mir222-enriched adsc-exosomes mitigate PM exposure-exacerbated cardiac ischemia-reperfusion injury through the modulation of the BNIP3-MAP1LC3B-BBC3/PUMA pathway. Autophagy. 2024;21:374-93.
14. Schütte JP, Manke M, Hemmen K, et al. Platelet-derived MicroRNAs regulate cardiac remodeling after myocardial ischemia. Circ Res. 2023;132:e96-113.
15. Senesi G, Lodrini AM, Mohammed S, et al. miR-24-3p secreted as extracellular vesicle cargo by cardiomyocytes inhibits fibrosis in human cardiac microtissues. Cardiovasc Res. 2025;121:143-56.
16. Kornieieva D, Kalocayova B, Slezak J, Kura B. Exploring the potential of molecular hydrogen in different heart failure models: a review. Int J Mol Sci. 2025;26:11574.
17. Qian L, Zhao Q, Yu P, et al. Diagnostic potential of a circulating miRNA model associated with therapeutic effect in heart failure. J Transl Med. 2022;20:267.
18. Zhang Z, Zou Z, Zhang H, Zhang D. Regulatory network analysis based on integrated miRNA-TF reveals key genes in heart failure. Sci Rep. 2024;14:13896.
19. Gomes CPDC, Schroen B, Kuster GM, et al. Regulatory RNAs in heart failure. Circulation. 2020;141:313-28.
20. Paterek A, Załęska-Kocięcka M, Surzykiewicz M, Wojdyńska Z, Leszek P, Mączewski M. Non-coding RNA therapeutics in the treatment of heart failure. Eur Heart J Cardiovasc Pharmacother. 2024;10:353-60.
21. Li W, Liu M, Zhao C, et al. MiR-1/133 attenuates cardiomyocyte apoptosis and electrical remodeling in mice with viral myocarditis. Cardiol J. 2020;27:285-94.
23. Shirakabe A, Ikeda Y, Uchikado Y, et al. Prognostic impact of mitochondrial dynamics-related miRNA levels during the treatment of acute heart failure in the hospital. Sci Rep. 2025;15:39938.
24. Larocca TJ, Seeger T, Prado M, et al. Pharmacological silencing of MicroRNA-152 prevents pressure overload-induced heart failure. Circ Heart Fail. 2020;13:e006298.
25. Shirakabe A, Ikeda Y, Uchikado Y, et al. Prognostic impact of excessive mitochondrial fission in patients with heart failure and evaluation of mitochondrial dynamics-related miRNAs in heart failure. Hypertens Res. 2025;48:2950-60.
26. Mohamed DI, Ezzat SF, Elayat WM, et al. Hepatoprotective role of carvedilol against ischemic hepatitis associated with acute heart failure via targeting miRNA-17 and mitochondrial dynamics-related proteins: an in vivo and in silico study. Pharmaceuticals. 2022;15:832.
27. Micu MA, Cozac DA, Scridon A. miRNA-orchestrated fibroinflammatory responses in heart failure with preserved ejection fraction: translational opportunities for precision medicine. Diagnostics. 2025;15:2286.
28. Wypijewska Del Nogal A, Sundar Rajan V, Westerlund F, Wilhelmsson LM. Complex conformational dynamics of the heart failure-associated Pre-miRNA-377 hairpin revealed by single-molecule optical tweezers. Int J Mol Sci. 2021;22:9008.
29. He Y, Dai MS, Tao LY, Gu X, Wang H, Liu P. Pericarpium trichosanthis inhibits TGF‐β1‐Smad3 pathway‐induced cardiac fibrosis in heart failure rats via upregulation of microRNA‐29b. J Gene Med. 2025;27:e70003.
30. Tao Y, Gao C, Qian D, Cao D, Han L, Yang L. Regulatory mechanism of fibrosis-related genes in patients with heart failure. Front Genet. 2022;13:1032572.
31. Gisa V, Islam MR, Lbik D, et al. Role of compensatory miRNA networks in cognitive recovery from heart failure. ncRNA. 2025;11:45.
32. Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376-85.
33. Kim H, Lee Y, Kim VN. The biogenesis and regulation of animal microRNAs. Nat Rev Mol Cell Biol. 2024;26:276-96.
35. Seida M, Ogami K, Yoshino S, Suzuki HI. Fine regulation of MicroRNAs in gene regulatory networks and pathophysiology. Int J Mol Sci. 2025;26:2861.
36. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99-110.
37. Bologna NG, Voinnet O. The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu Rev Plant Biol. 2014;65:473-503.
38. Schirle NT, Sheu-Gruttadauria J, Macrae IJ. Structural basis for microRNA targeting. Science. 2014;346:608-13.
39. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421-33.
40. Luisotti L, Germelli L, Piccarducci R, Giacomelli C, Marchetti L, Martini C. Extracellular vesicles as vehicles for small non-coding RNA therapeutics: standardization challenges for clinical translation. Extracell Vesicles Circ Nucleic Acids. 2025;6:398-427.
41. Akbar N, Azzimato V, Choudhury RP, Aouadi M. Extracellular vesicles in metabolic disease. Diabetologia. 2019;62:2179-87.
42. Huang W, Zhang Q, Qi H, et al. Deletion of neuropeptide Y attenuates cardiac dysfunction and apoptosis during acute myocardial infarction. Front Pharmacol. 2019;10:1268.
43. Sun T, Dong C, Xiong S. Cardiomyocyte‐derived HMGB1 takes a protective role in CVB3‐induced viral myocarditis via inhibiting cardiac apoptosis. Immunol Cell Biol. 2023;101:735-45.
44. Hao H, Yuan T, Li Z, et al. Curcumin analogue C66 ameliorates mouse cardiac dysfunction and structural disorders after acute myocardial infarction via suppressing JNK activation. Eur J Pharmacol. 2023;946:175629.
45. Mentkowski KI, Tarvirdizadeh T, Manzanero CA, Eagler LA, Lang JK. Surface engineering enhances the therapeutic potential of systemically delivered extracellular vesicles following acute myocardial infarction. FASEB J. 2024;38:e70070.
46. Lu C, Chen D, Dong K, et al. Inhibition of miR-143-3p alleviates myocardial ischemia reperfusion injury via limiting mitochondria-mediated apoptosis. Biol Chem. 2023;404:619-31.
47. Liao B, Dong S, Xu Z, Gao F, Zhang S, Liang R. MiR-19b-3p regulated by BC002059/ABHD10 axis promotes cell apoptosis in myocardial infarction. Biol Direct. 2022;17:20.
48. Zhang Q, Yu N, Yu BT. MicroRNA-298 regulates apoptosis of cardiomyocytes after myocardial infarction. Eur Rev Med Pharmacol Sci. 2018;22:532-9.
49. Wang Z, Wei TYW, Chen JMM, et al. MicroRNA‐100‐5p exacerbates myocardial ischemia-reperfusion injury through downregulation of PRMT5. FASEB J. 2025;39:e70690.
50. Xu C, Hu Y, Hou L, et al. β-Blocker carvedilol protects cardiomyocytes against oxidative stress-induced apoptosis by up-regulating miR-133 expression. J Mol Cell Cardiol. 2014;75:111-21.
51. Zong L, Wang W. CircANXA2 promotes myocardial apoptosis in myocardial ischemia-reperfusion injury via inhibiting miRNA‐133 expression. BioMed Res Int. 2020;2020:8590861.
52. Zhang J, Luo CJ, Xiong XQ, et al. MiR-21-5p-expressing bone marrow mesenchymal stem cells alleviate myocardial ischemia/reperfusion injury by regulating the circRNA_0031672/miR-21-5p/programmed cell death protein 4 pathway. J Geriatr Cardiol. 2021;18:1029-43.
53. Xiao J, Pan Y, Li XH, et al. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis. 2016;7:e2277.
54. Olson JM, Yan Y, Bai X, et al. Up-regulation of MicroRNA-21 mediates isoflurane-induced protection of cardiomyocytes. Anesthesiology. 2015;122:795-805.
55. Tang M, Pan H, Zheng Z, et al. Prostaglandin E1 protects cardiomyocytes against hypoxia-reperfusion induced injury via the miR-21-5p/FASLG axis. Biosci Rep. 2019;39:BSR20190597.
56. Wang Q, Xie Z. GAS5 silencing attenuates hypoxia‐induced cardiomyocytes injury by targeting miR‐21/PTEN. Immun Inflamm Dis. 2023;11:e945.
57. Liu Z, Wang H, Hou G, Cao H, Zhao Y, Yang B. Notoginsenoside R1 protects oxygen and glucose deprivation‐induced injury by upregulation of miR‐21 in cardiomyocytes. J Cell Biochem. 2018;120:9181-92.
58. Lv X, Lu P, Hu Y, Xu T. miR-346 inhibited apoptosis against myocardial ischemia-reperfusion injury via targeting bax in rats. Drug Des Devel Ther. 2020;14:895-905.
59. Jia Z, Wang J, Shi Q, et al. SOX6 and PDCD4 enhance cardiomyocyte apoptosis through LPS-induced miR-499 inhibition. Apoptosis. 2015;21:174-83.
60. Yang G, Tang X, Tan L, Nong D, Yang P, Ning H. Upregulation of miR-144-3p protects myocardial function from ischemia-reperfusion injury through inhibition of TMEM16A Ca2+-activated chloride channel. Hum Cell. 2021;34:360-71.
61. Zhao Z, Qu F, Liu R, Xia Y. Differential expression of miR‐142‐3p protects cardiomyocytes from myocardial ischemia‐reperfusion via TLR4/NFkB axis. J Cell Biochem. 2019;121:3679-90.
62. Zhu H, Wang X, Sun Y, et al. MicroRNA-486-5p targeting PTEN protects against coronary microembolization-induced cardiomyocyte apoptosis in rats by activating the PI3K/AKT pathway. Eur J Pharmacol. 2019;855:244-51.
63. Wang C, Zhang C, Liu L, et al. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther. 2017;25:192-204.
64. Yang L, Dai Q, Bao X, Li W, Liu J. MiR-4763-3p accelerates lipopolysaccharide-induced cardiomyocyte apoptosis and inflammatory response by targeting IL10RA. Cytotechnology. 2023;76:179-90.
65. Xu M, Li X, Song L, Tao C, Fang J, Tao L. miR-484 targeting of Yap1-induced LPS-inhibited proliferation, and promoted apoptosis and inflammation in cardiomyocyte. Biosci Biotechnol Biochem. 2021;85:378-85.
66. Guo X, Ji Q, Wu M, Ma W. Naringin attenuates acute myocardial ischemia-reperfusion injury via miR- 126/GSK-3β/β-catenin signaling pathway. Acta Cir Bras. 2022;37:e370102.
67. Corsten MF, Heggermont W, Papageorgiou A, et al. The microRNA-221/-222 cluster balances the antiviral and inflammatory response in viral myocarditis. Eur Heart J. 2015;36:2909-19.
68. Ma C, Yang Z, Wang J, et al. Interleukin-1β-stimulated macrophage-derived exosomes improve myocardial injury in sepsis via regulation of mitochondrial homeostasis: experimental research. Int J Surg. 2025;111:283-301.
69. Diniz GP, Lino CA, Moreno CR, Senger N, Barreto‐Chaves MLM. MicroRNA‐1 overexpression blunts cardiomyocyte hypertrophy elicited by thyroid hormone. J Cell Physiol. 2017;232:3360-8.
70. Li Q, Song X, Zou J, et al. Attenuation of microRNA-1 derepresses the cytoskeleton regulatory protein twinfilin-1 to provoke cardiac hypertrophy. J Cell Sci. 2010;123:2444-52.
71. Diniz GP, Lino CA, Guedes EC, Nascimento Moreira LD, Barreto-Chaves MLM. Cardiac microRNA-133 is down-regulated in thyroid hormone-mediated cardiac hypertrophy partially via Type 1 Angiotensin II receptor. Basic Res Cardiol. 2015;110:49.
72. Liu X, Li H, Hastings MH, et al. miR-222 inhibits pathological cardiac hypertrophy and heart failure. Cardiovasc Res. 2024;120:262-72.
73. Zhao L, Qian X, Ren Z, Wang A. miR‐31‐5p suppresses myocardial hypertrophy by targeting Nfatc2ip. J Cell Mol Med. 2024;28:e18413.
74. Li J, Sha Z, Zhu X, et al. Targeting miR-30d reverses pathological cardiac hypertrophy. eBioMedicine. 2022;81:104108.
75. Huang X, Li J, Li X, et al. miR-208a in cardiac hypertrophy and remodeling. Front Cardiovasc Med. 2021;8:773314.
76. Heymans S, Corsten MF, Verhesen W, et al. Macrophage MicroRNA-155 promotes cardiac hypertrophy and failure. Circulation. 2013;128:1420-32.
77. Zhang Y, Ding Y, Li M, et al. MicroRNA-34c-5p provokes isoprenaline-induced cardiac hypertrophy by modulating autophagy via targeting ATG4B. Acta Pharm Sin B. 2022;12:2374-90.
78. Yu S, Wang M, Xie Y, Wang B, Xu Y. MiR-495-3p promotes cardiac hypertrophy by targeting Pum2. Cell Mol Biol. 2024;70:116-20.
79. Long M, Cheng M. Small extracellular vesicles associated miRNA in myocardial fibrosis. Biochem Biophys Res Commun. 2024;727:150336.
80. Liu B, Wei Y, He J, et al. Human umbilical cord-derived mesenchymal stromal cells improve myocardial fibrosis and restore miRNA-133a expression in diabetic cardiomyopathy. Stem Cell Res Ther. 2024;15:120.
81. Zhang SL, Fan FL, Wei F, Wang J, Zhang YS. Effect of microRNA-133b on myocardial fibrosis. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2019;41:589-94.
82. Guo F, Tang C, Huang B, et al. LncRNA H19 drives proliferation of cardiac fibroblasts and collagen production via suppression of the miR-29a-3p/miR-29b-3p-VEGFA/TGF-β axis. Mol Cells. 2022;45:122-33.
83. Yuan J, Yang H, Liu C, et al. Microneedle patch loaded with exosomes containing MicroRNA‐29b prevents cardiac fibrosis after myocardial infarction. Adv Healthc Mater. 2023;12:2202959.
84. Wang Y, Yu J, Ou C, et al. miRNA-146a-5p inhibits hypoxia-induced myocardial fibrosis through EndMT. Cardiovasc Toxicol. 2024;24:133-45.
85. Huang D, Wen Q, Su Y, Li X. miR-17-5p inhibits BNIP3-mediated mitochondrial autophagy to attenuate pathological cardiac fibrosis. Balkan Med J. 2025;42:516-25.
86. Zhao H, Yang H, Geng C, et al. Elevated IgE promotes cardiac fibrosis by suppressing miR-486a-5p. Theranostics. 2021;11:7600-15.
87. Xia YW, Wang SB. Microvesicles containing microRNA-21 induce myocardial fibrosis via AKT pathway. Eur Rev Med Pharmacol Sci. 2018;22:4634-41.
88. Yuan J, Chen H, Ge D, et al. Mir-21 promotes cardiac fibrosis after myocardial infarction via targeting Smad7. Cell Physiol Biochem. 2017;42:2207-19.
89. Yang J, Yu X, Xue F, et al. Exosomes derived from cardiomyocytes promote cardiac fibrosis via myocyte-fibroblast cross-talk. Am J Transl Res. 2018;10:4350-66.
90. Cao F, Li Z, Ding W, Qv C, Zhao H. Exosomal miR-15a-5p from cardiomyocytes promotes myocardial fibrosis. Mol Cell Biochem. 2024;480:1701-13.
91. Wu H, Zhuang Y, Yue Y, et al. SPOP is a key trigger of pathological cardiac hypertrophy and heart failure. Circ Res. 2025;137:e177-96.
92. Jebran AF, Seidler T, Tiburcy M, et al. Engineered heart muscle allografts for heart repair in primates and humans. Nature. 2025;639:503-11.
93. Chen X, Yu X, Zhong S, et al. ALDH2/eIF3E interaction modulates protein translation critical for cardiomyocyte ferroptosis in acute myocardial ischemia injury. Circulation. 2026;153:164-84.
94. Ciccarelli M, Pires IF, Bauersachs J, et al. Acute heart failure: mechanisms and pre-clinical models—a scientific statement of the ESC working group on myocardial function. Cardiovasc Res. 2023;119:2390-404.
95. Li Q, Yao Y, Shi S, et al. Inhibition of miR‐21 alleviated cardiac perivascular fibrosis via repressing EndMT in T1DM. J Cell Mol Med. 2019;24:910-20.
96. Zhang Y, Yuan B, Xu Y, et al. MiR-208b/miR-21 promotes the progression of cardiac fibrosis through the activation of the TGF-β1/Smad-3 signaling pathway: an in vitro and in vivo study. Front Cardiovasc Med. 2022;9:924629.
97. Wang X, Zhang T, Zhai J, et al. MiR-21 attenuates FAS-mediated cardiomyocyte apoptosis by regulating HIPK3 expression. Biosci Rep. 2023;43:BSR20230014.
98. Holland A, Enrick M, Diaz A, Yin L. Is miR-21 a therapeutic target in cardiovascular disease? Int J Drug Discov Pharm. 2023;2:26-36.
99. Gozdowska R, Makowska A, Gąsecka A, Chabior A, Marchel M. Circulating microRNA in heart failure—practical guidebook to clinical application. Cardiol Rev. 2020;30:16-23.
100. Fathy WM, Montaser BA, Ibrahim WA, Salah DM, Aboelkhair NT. The role of microRNA-19b, microRNA-21, and microRNA-208a in diagnosis of heart failure. BMC Cardiovasc Disord. 2025;25:864.
101. Wong LL, Zou R, Zhou L, et al. Combining circulating MicroRNA and NT-proBNP to detect and categorize heart failure subtypes. J Am Coll Cardiol. 2019;73:1300-13.
102. Gallo A, Agnese V, Sciacca S, et al. MicroRNA-30d and -483-3p for Bi-ventricular remodelling and miR-126-3p for pulmonary hypertension in advanced heart failure. ESC Heart Fail. 2024;11:155-66.
103. Tong L, Wang Q, Zhang Y, et al. Myocardial delivery of miR30d with peptide-functionalized milk-derived extracellular vesicles for targeted treatment of hypertrophic heart failure. Biomaterials. 2025;316:122976.
104. Foinquinos A, Batkai S, Genschel C, et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat Commun. 2020;11:633.
105. Täubel J, Hauke W, Rump S, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. 2021;42:178-88.
106. Zhou H, Tang W, Yang J, Peng J, Guo J, Fan C. MicroRNA-related strategies to improve cardiac function in heart failure. Front Cardiovasc Med. 2021;8:773083.
107. Forterre A, Komuro H, Aminova S, Harada M. A Comprehensive review of cancer MicroRNA therapeutic delivery strategies. Cancers. 2020;12:1852.
108. Wu Q, Yao X, Duan X, Chen X, Zhang J. Nanomedicine reimagined: translational strategies for precision tumor theranostics. Adv Mater. 2025;38:e10293.
109. Li X, Chen Y, Cao X, Feng W, Chen Y, Zhang J. Inflammatory macrophage-targeted atherosclerosis treatment by miRNA-delivered, MRI-visible, and anti-inflammatory nanomedicine. ACS Nano. 2025;19:20472-90.
110. Wang Z, Chen J, Wang J, et al. MSCs biomimetic ultrasonic phase change nanoparticles promotes cardiac functional recovery after acute myocardial infarction. Biomaterials. 2025;313:122775.
111. Nazari-shafti TZ, Exarchos V, Biefer HRC, et al. MicroRNA mediated cardioprotection - is there a path to clinical translation? Front Bioeng Biotechnol. 2020;8:149.
112. Gao L, Qiu F, Cao H, et al. Therapeutic delivery of microRNA-125a-5p oligonucleotides improves recovery from myocardial ischemia/reperfusion injury in mice and swine. Theranostics. 2023;13:685-703.
113. Chen Y, Liu S, Liang Y, et al. Single dose of intravenous miR199a-5p delivery targeting ischemic heart for long-term repair of myocardial infarction. Nat Commun. 2024;15:5565.
114. Baek ST, Kerjan G, Bielas SL, et al. Off-target effect of doublecortin family shRNA on neuronal migration associated with endogenous MicroRNA dysregulation. Neuron. 2014;82:1255-62.
115. Asensio Lopez MDC, Ruiz Ballester M, Bastida Nicolas FJ, et al. miR-106b-5p as a central regulator of cancer progression and chemotherapy-induced cardiotoxicity: from molecular mechanisms to clinical translation. Int J Mol Sci. 2025;26:10002.
116. Chen X, Chen H, Zhu L, et al. Nanoparticle-patch system for localized, effective, and sustained miRNA Administration into infarcted myocardium to alleviate myocardial ischemia-reperfusion injury. ACS Nano. 2024;18:19470-88.
117. Wang Y, Meng D, Shi X, et al. Injectable hydrogel with miR-222-engineered extracellular vesicles ameliorates myocardial ischemic reperfusion injury via mechanotransduction. Cell Rep Med. 2025;6:101987.
118. Hong DS, Kang Y, Borad M, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. 2020;122:1630-7.
119. Li W, Wang Y, Liu R, et al. MicroRNA-34a: potent tumor suppressor, cancer stem cell inhibitor, and potential anticancer therapeutic. Front Cell Dev Biol. 2021;9:640587.
120. Lv P, Zhou M, He J, et al. Circulating miR-208b and miR-34a are associated with left ventricular remodeling after acute myocardial infarction. Int J Mol Sci. 2014;15:5774-88.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.