


Emerging roles and mechanisms of small nucleolar RNA in cardiovascular diseases: research progress
 0
0 Abstract
Small nucleolar RNAs (snoRNAs) are increasingly recognized as key regulatory factors in cardiovascular disease (CVD), with functions that extend far beyond the traditional scope of ribosomal RNA modification. This review synthesizes current knowledge regarding the biology of snoRNAs and their emerging roles in CVD, aiming to provide a clear theoretical framework for their pathogenic mechanisms and clinical significance. We first discuss the biogenesis, classification, and functional diversity of snoRNAs, covering both classical modification roles and non-classical functions such as the regulation of messenger RNA splicing, participation in the DNA damage response, and the generation of snoRNA-derived small RNAs. In CVD, snoRNAs exhibit spatiotemporal dysregulation and are extensively involved in pathological processes ranging from congenital heart defects and cardiomyopathy to coronary artery disease, myocardial infarction, arrhythmias, and heart failure. Specific gene clusters, particularly 14q32 snoRNAs, repeatedly emerge as key regulatory nodes in processes such as vascular remodeling and platelet activation. However, the majority of dysregulated snoRNAs remain orphaned with unknown targets, limiting our understanding of their mechanisms and clinical translation. We emphasize that combining computational modeling with multi-omics approaches can accelerate target identification and functional elucidation. Finally, we systematically examine the opportunities and challenges facing the clinical translation of snoRNA research, including their potential as dynamic biomarkers, limitations of detection technologies, and barriers to therapeutic delivery. By comprehensively reviewing existing evidence and future directions, this review highlights the significant potential of snoRNAs as novel biomarkers and therapeutic targets for the precision diagnosis and treatment of cardiovascular diseases.
Keywords
INTRODUCTION
Cardiovascular disease (CVD) is a leading cause of morbidity and mortality worldwide. According to data published by the Global Burden of Disease, by 2023, the global number of disability-adjusted life years attributable to CVD reached 437 million (95% uncertainty interval (UI): 401-465 million), with 19.2 million deaths (95% UI: 17.4-20.4 million)[1]. Despite improvements in public health literacy and advances in medical interventions and technology, which have successfully curbed certain risk factors like smoking and reduced CVD mortality, the burden of CVD continues to escalate. This is driven by unhealthy diets, obesity, elevated fasting blood glucose, low physical activity, and population aging, posing significant threats to human society[1,2]. Moreover, the widespread adoption of precision medicine and the application of biologics have significantly advanced the comprehensive management of CVD[3,4]. Nevertheless, limitations in understanding the pathophysiology of CVD, particularly the underlying mechanisms of gene regulation, have made identifying and validating novel therapeutic targets exceptionally challenging. This has become a critical bottleneck constraining clinical translation in this field[5,6]. Therefore, there is an urgent need to map the genetic landscape of CVD, uncover potential biomarkers, and identify novel therapeutic targets. In this exploration, the research focus is expanding from traditional protein-coding genes to the non-coding regions that constitute the vast majority of the genome.
Non-coding RNA (ncRNA) increasingly plays a central regulatory role in cardiac development and disease, participating in numerous pathological processes through precise gene regulation[7,8]. Among these, small nucleolar RNAs (snoRNAs) are emerging as a promising research focus due to their unique functions in post-transcriptional modifications and novel regulatory mechanisms[9]. snoRNAs are a class of short RNAs, typically 60-300 nucleotides in length, primarily localized within the nucleoli of eukaryotic cells. The vast majority (approximately 90%) originate from intron regions of coding genes[10]. Their maturation process relies on the precise processing of their precursor transcripts, specifically through intron splicing or cleavage by specific nucleases to remove the “cap” and “polyadenylate tail” structures at both ends[11]. Traditionally, snoRNAs have been regarded as housekeeping RNA molecules whose primary function is to direct RNA chemical modifications, including 2'-O-methylation and pseudouridylation, which are crucial for proper ribosomal assembly and efficient protein translation[12,13]. However, recent studies have revealed that they can also mediate ribosomal RNA (rRNA) acetylation and transfer RNA (tRNA) methylation, regulate splicing patterns and messenger RNA (mRNA) abundance, and form various RNA derivatives[14]. This functional diversity implies a more complex role for snoRNAs in cellular and tissue homeostasis. Notably, mounting evidence indicates that snoRNAs exhibit spatiotemporal specificity in cardiac tissue, with their abnormal expression significantly associated with the onset and progression of various diseases including myocardial infarction (MI), heart failure (HF), and atherosclerosis (AS)[15,16]. For instance, analysis of whole-genome data from 5,244 participants in the Prospective Study of Elderly Patients on Pravastatin (PROSPER) revealed that 14q32 snoRNAs expression exhibits high vascular specificity. This snoRNA binds to the methyltransferase fibrillarin and participates in the pathological process of heart failure[17]. Notably, measuring circulating 14q32 snoRNAs levels in peripheral artery disease patients revealed four abnormally expressed snoRNAs, with SNORD113-2 and SNORD114-1 levels closely linked to platelet activation[16]. These findings strongly suggest snoRNAs may represent an important, yet poorly understood, component of the CVD gene regulatory network.
However, our understanding of the role of snoRNAs in CVD remains highly fragmented and limited. Most studies have only reached the stage of differential expression analysis, with the cell types involved, underlying molecular mechanisms, and regulated biological targets largely unknown. This knowledge gap significantly impedes our progress in utilizing snoRNAs as future therapeutic targets and biomarkers for CVD. Therefore, this review aims to systematically summarize current research progress on snoRNAs in CVD. We will first elaborate on the biogenesis and functional diversity of snoRNAs; subsequently, we will delve into their functional roles in CVD; finally, we will focus on the latest technological advances in snoRNA research to identify the biological functions of more orphan snoRNAs.
CLASSIFICATION AND CHARACTERISTICS OF snoRNAs
C/D box snoRNAs
C/D box snoRNAs typically range in length from 50 to 100 nucleotides and are characterized by two conserved sequences near the RNA terminus: the C box (RUGAUGA) and the D box (CUGA)[18]. The G-A base pairs within the C and D boxes, formed via sugar-edge Hoogsteen pairing, induce a sharp bend in the otherwise linear RNA chain, forming a characteristic kink-turn (k-turn) motif[19]. Using the k-turn as a scaffold, they bind to core proteins (including SNU13, NOP56, NOP58, and fibrillarin) to form a catalytically active small nucleolar ribonucleoprotein (snoRNP) complex [Figure 1][20]. This complex recognizes and binds specific regions of rRNA through base-pairing between the antisense element (ASE), which comprises the single-stranded sequences positioned between the C and D' boxes and between the C' and D boxes, and the target rRNA sequence. In this process, the nucleotide distance between the D box and ASE determines targeting accuracy, while the nucleotide sequence of ASE itself determines the specific rRNA and site recognized[21]. In recent years, with the discovery of non-canonical functions of C/D box snoRNAs regulating chromatin compaction, metabolic stress, cholesterol transport, alternative splicing, and mRNA levels, the complementarity of the ASE is no longer the sole criterion for identifying potential targets[22].
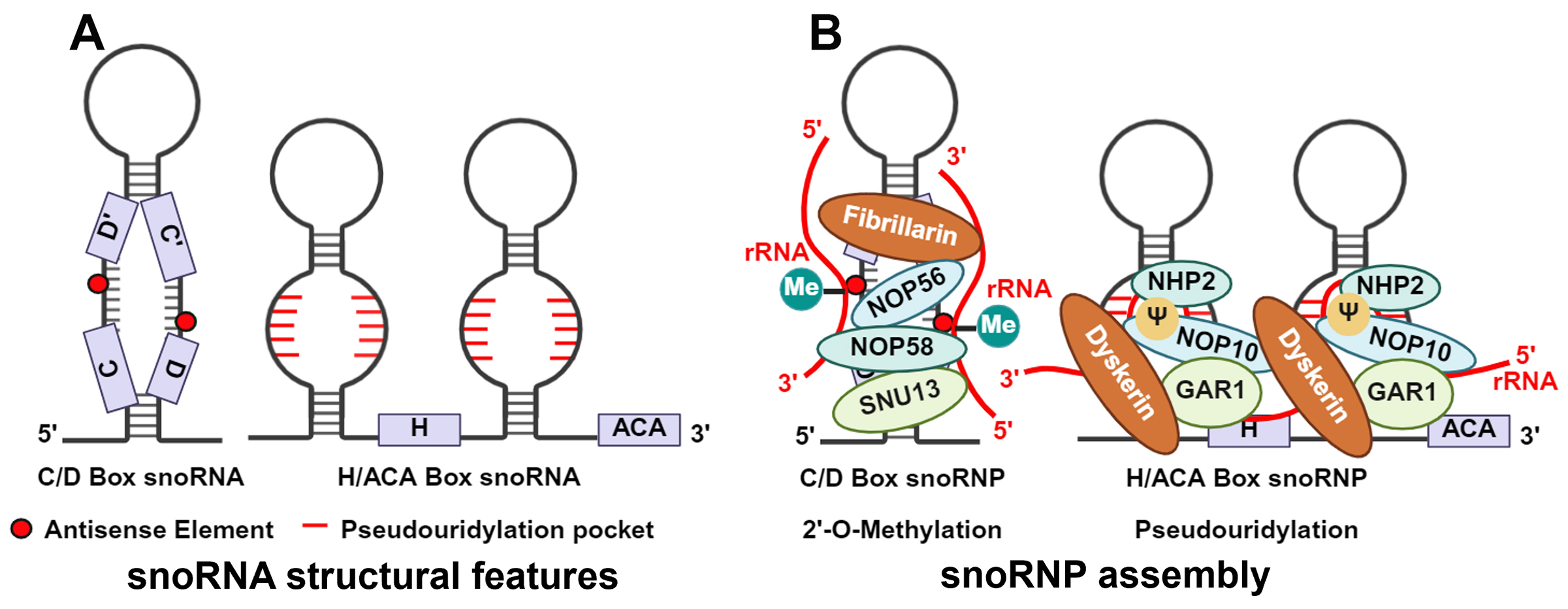
Figure 1. Schematic of snoRNA structure and snoRNP assembly. (A) snoRNA structural features. (B) snoRNP assembly. The C/D box snoRNP is composed of a snoRNA bound to four core proteins—SNU13, NOP58, NOP56, and Fibrillarin—and catalyzes site-specific 2'-O-ribose methylation of target RNAs through its antisense element. The H/ACA box snoRNP consists of a snoRNA associated with the four core proteins GAR1, NOP10, NHP2, and Dyskerin, and facilitates pseudouridylation of target RNAs via its pseudouridylation pocket. (Created in BioRender. Cao, H. (2026) https://BioRender.com/o1aj8wx).
H/ACA box snoRNAs
H/ACA box snoRNAs range in length from 120 to 140 nucleotides and are characterized by a highly conserved “hairpin-hinge-hairpin-tail” secondary structure. The H box (ANANNA) and ACA box are located near the hairpin in the hinge and tail regions, respectively[23]. The ACA box is essential for recruiting and stably binding the core proteins, directly affecting the efficiency of pseudouridylation function[24]. H/ACA box snoRNAs lack k-turn structures but recruit Dyskerin, NOP10, NHP2, and GAR1 proteins to form snoRNP [Figure 1]. Dyskerin serves as the catalytic subunit. The specificity of the reaction is determined by two short, complementary sequences (9-16 nucleotides) within the internal loops of the hairpins, which form the "pseudouridylation pockets" that base-pair with the target RNA and position the modification site into Dyskerin's active site[23,25].
scaRNAs
Small Cajal body-specific RNAs (scaRNAs) are a class of small RNAs specific to Cajal bodies, primarily localized to these nuclear structures via UG repeat sequences[26]. Its structure exhibits features characteristic of both C/D box snoRNAs and H/ACA box snoRNAs, thereby mediating 2'-O-methylation and pseudouridylation of target small nuclear RNAs (snRNAs). Recent studies indicate that under conditions of transactivation response element-binding protein 43 (TDP-43) depletion, scaRNAs translocate from Cajal bodies to the nucleolus. This translocation occurs because TDP-43 deficiency impairs the ability of the Cajal body anchor protein WD40-repeat protein 79 to bind specific scaRNAs. This mislocalization ultimately impairs their function in mediating the 2'-O-methylation of snRNAs[26].
slb-snoRNAs, lnc-snoRNAs and orphan snoRNAs
Short loop box snoRNAs (slb-snoRNAs) are a class of snoRNAs similar to C/D box snoRNAs, lacking only the internal C'/D' box loop structure, yet their function is indistinguishable from that of canonical C/D box snoRNAs[27,28]. snoRNA-related long non-coding RNAs (lnc-snoRNAs) constitute a group of ncRNAs localized within the Prader-Willi syndrome region of human chromosome 15. These are essentially long non-coding RNAs (lncRNAs) flanked by C/D box or H/ACA box snoRNAs at both ends. It has been reported that the C-box element in the 5'-terminal snoRNAs and the D-box motif in the 3'-terminal snoRNAs are essential for their proper processing. This unique structure enables the snoRNAs to protect the lncRNA precursor, which lacks a typical 5'-cap and 3'-polyadenylation tail, thereby maintaining its stability[29,30]. Furthermore, snoRNAs with unknown targets are termed orphan snoRNAs[31]. Identifying the targets of these orphan snoRNAs is therefore crucial for elucidating their physiological roles and understanding their contributions to disease.
THE CLASSIC PHYSIOLOGICAL FUNCTIONS OF snoRNAs
As described above, traditionally, C/D box snoRNAs primarily promote 2′-O-methylation of rRNA, while H/ACA box snoRNAs direct pseudouridylation of rRNA [Figure 2]. In fact, the ASE of certain snoRNAs shows no similarity to known rRNA 2′-O-methylation sites, suggesting their potential role in directing this modification to other classes of RNA[32]. Fibrillarin is the only known snoRNP-dependent 2'-O-methyltransferase that mediates snoRNA-directed modifications of mRNA or tRNA[33]. For example, Fibrillarin-mediated 2'-O-methylation is associated with widespread 3' untranslated region (UTR) truncation of mRNA, a modification strategy that regulates gene expression by enhancing mRNA stability[34]. Concurrently, earlier studies demonstrated that snoRNPs carrying SNORD97 and SCARNA97, respectively, mediate 2'-O-methylation of cytidine at position C34 of the tRNAMet swing site in human elongator factors, a process protecting tRNA from stress-induced ribonuclease cleavage[35]. Recent systematic evidence indicates that in human HEK293, the SNORD97-containing snoRNP serves as a pivotal node in the global tRNA 2'-O-methylation network, regulating cellular proliferation and differentiation state transitions by controlling methionine utilization[36]. Through these mechanisms, C/D box snoRNAs alter the spatial structure of ribosomes and mRNAs while regulating their stability, thereby controlling cellular transcription levels and providing critical guidance for subsequent translation. Similarly, the role of H/ACA box snoRNAs in mRNA and tRNA pseudouridylation modifications has been extensively validated. A frontier advancement in this field is the ability to artificially design and synthesize H/ACA snoRNAs, enabling programmed manipulation of downstream target RNA expression levels. Specifically, designing H/ACA guide RNAs that target premature termination codons on mRNAs induces pseudouridine (Ψ) modification formation[37]. This strategy was explicitly validated in Pan et al.'s study: they discovered that this modification forms a unique Ψ-Ψ codon-anticodon pairing at premature termination codons, significantly enhancing read-through efficiency and enabling precise reprogramming of the translation process[38]. Additionally, Ψ can stabilize adjacent nucleotides, potentially affecting ribosomal fidelity[39].
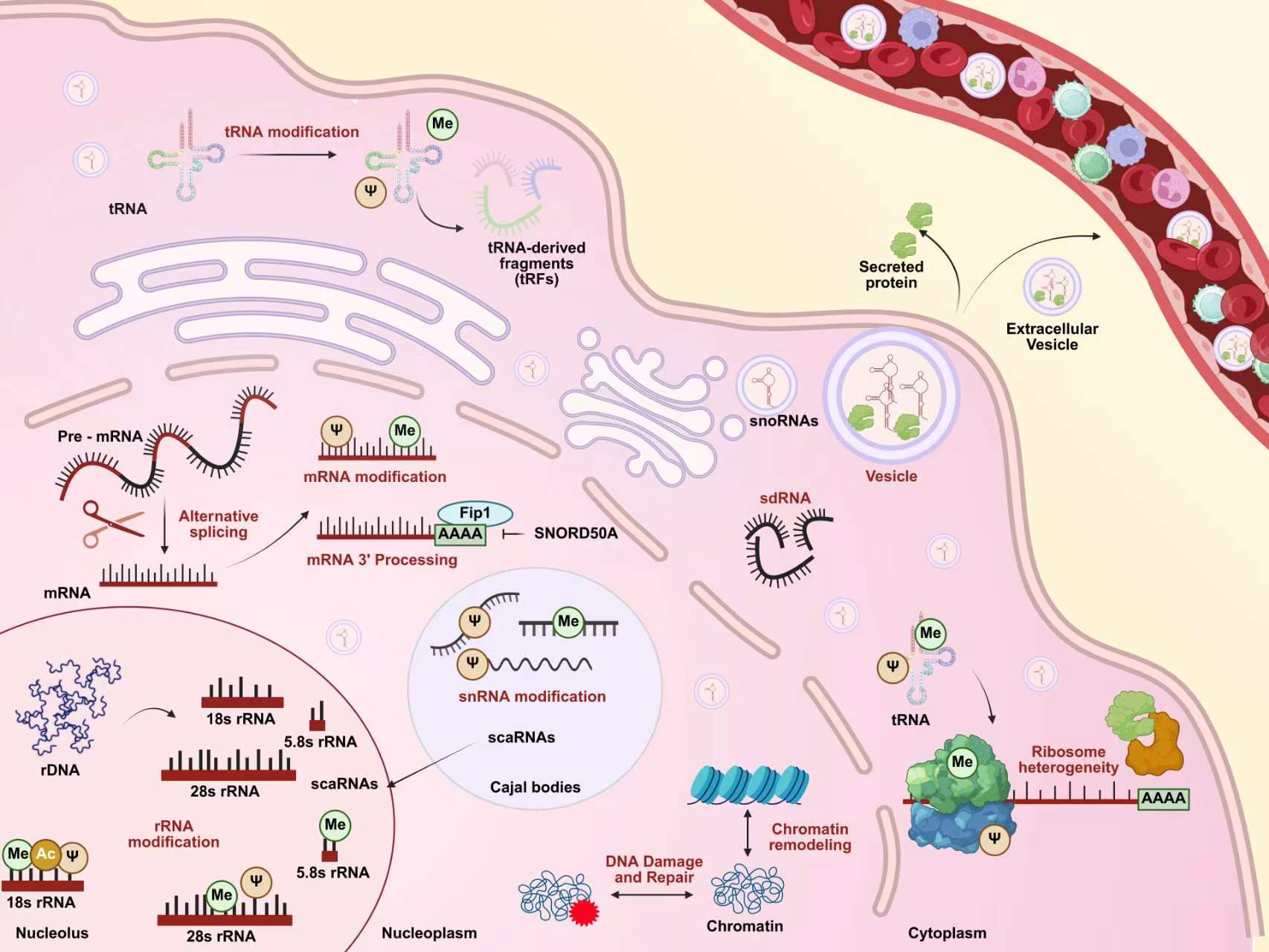
Figure 2. Biological functions of snoRNAs. Within the nucleolus, snoRNAs primarily guide the processing, modification, and maturation of ribosomal RNA (rRNA), including site-specific 2'-O-methylation, pseudouridylation, and acetylation. In the nucleoplasm, beyond their roles in pre-mRNA processing, alternative splicing, 3'-end formation, and chemical modification of transcripts, snoRNAs are involved in key nuclear processes such as chromatin remodeling and DNA damage repair. Small Cajal body-specific RNAs (scaRNAs), which modify small nuclear RNAs (snRNAs), are localized to Cajal bodies and can translocate to the nucleolus under specific conditions. Furthermore, snoRNAs can be processed into smaller snoRNA-derived RNAs (sdRNAs) that enter the cytoplasm, where they regulate transfer RNA (tRNA) modifications, are packaged into extracellular vesicles for secretion into the circulatory system, and contribute to ribosome heterogeneity. This highlights the multifaceted roles of snoRNAs, spanning from nuclear RNA processing to extracellular signaling. (Created in BioRender. Cao, H. (2026) https://BioRender.com/mc5sxut).
This evidence suggests that snoRNAs play a crucial role in the fine-tuning of protein synthesis by guiding specific modifications of rRNA, tRNA, and mRNA.
NON-CANONICAL PHYSIOLOGICAL FUNCTIONS OF snoRNAs
In recent years, in-depth research has revealed that the functional landscape of snoRNAs extends far beyond their classical base-modification roles. Beyond these modification-based functions, snoRNAs have demonstrated broader non-classical activities, expanding their regulatory scope from the translation machinery itself to various levels of cellular physiology and even disease progression. These non-canonical functions—including directing acetylation modifications of 18S rRNA, regulating mRNA processing and alternative splicing, deriving into functionally active snoRNA-derived RNAs (sdRNAs), and even participating in DNA damage responses—unveil snoRNAs' novel identity as fine-tuners of cellular regulation. For CVD, this deepening understanding holds particularly critical significance. As an organ that beats throughout life, cardiac function relies heavily on precise gene expression and efficient protein machinery. Vascular endothelial cells require continuous adaptive responses to blood flow shear stress and metabolic stress[40]. Therefore, gaining a deeper understanding of how snoRNAs participate in these processes through the aforementioned non-canonical pathways will provide a novel perspective for elucidating the molecular mechanisms of cardiovascular diseases.
snoRNAs regulate acetylation of 18S rRNA
N-Acetyltransferase 10 (NAT10) is the sole known writer enzyme for N4-acetylcytidine (ac4C), and its catalytic activity can be potentiated through protein engineering[41,42]. In eukaryotes, a4C pairs located at helix 34 and helix 45 of 18S rRNA are crucial for maintaining translation fidelity[43,44]. In yeast, this modification is mediated by the orphan C/D box snoRNAs snR4 and snR45[45]. This mechanism is supported by cross-linking and hybridization sequencing data, which captured direct duplex formation between these snoRNAs and the target regions flanking the acetylation sites in rRNA[46]. Further studies indicate this mechanism is evolutionarily conserved. In humans, Lu et al. identified interaction between SNORD13, an ortholog of snR45, and 18S rRNA using Psoralen Analysis of RNA Interactions and Structures[47]. Subsequent functional studies first established that SNORD13 is essential for NAT10 to catalyze the ac4C modification at position C1842 on helix 45 of 18S rRNA[48]. Surprisingly, however, this mechanism exhibits significant variation across species. SNORD13 does not mediate ac4C modification in Caenorhabditis elegans, while Drosophila melanogaster employs an atypical SNORD13 containing only an antisense sequence, rather than the bifurcated structure used in humans[48,49]. These studies demonstrate that the regulatory networks formed between snoRNAs and their targets evolve dynamically, with both function and structure adapting to the physiological demands of different species. Therefore, elucidating the differences in rRNA acetylation mechanisms among species is crucial for understanding snoRNAs evolution and will greatly enrich our knowledge of the complex landscape of the eukaryotic RNA acetylation.
Meanwhile, these species-specific differences underscore the need for caution when extrapolating findings from model organisms to human CVD, highlighting the value of studies using human cell lines and clinical samples.
snoRNAs participate in mRNA processing and alternative splicing
The processing and maturation of mRNA are crucial for maintaining normal cellular physiological functions. As mentioned above, the vast majority of snoRNAs are generated through intron splicing of host mRNAs[10]. Disruption of this critical step leads to abnormal snoRNA maturation and loss of function in host mRNA transcripts. Liu et al. first elucidated the specific mechanism of this process: when splicing is inhibited, an erroneously amplified RNA accumulates extensively within cells, termed hybrid mRNA-snoRNAs (hmsnoRNAs)[50]. This molecule is generated by the nuclear spliceosome through sequential cleavages at the 3' end of pre-mRNA. It incorporates the 5' UTR of the host mRNA, the first exon, the upstream region of the intron, the complete snoRNA sequence, and a 3' end identical to that of mature snoRNAs. Subsequently, these hmsnoRNAs are transported to the cytoplasm, where they are cleared by Xrn1p via the standard mRNA degradation pathway[50]. This study demonstrates that snoRNA maturation is tightly coupled with mRNA 3'-end processing. For instance, the Integrator complex (whose key subunits are INTS9/INTS11), responsible for processing multiple RNA 3'-ends, exhibits impaired function upon BRAT1 deficiency. This defect simultaneously causes dysfunction in snoRNA 3'-end processing and impaired processing of replication-dependent histone pre-mRNAs[51]. Concurrently, mRNA 3'-end exons recruit the RNase III family enzyme Pac1, which cleaves downstream snoRNA precursors to promote their maturation[11]. More directly, Huang et al. demonstrated that U/A-rich SNORD50A inhibits mRNA 3'-end processing by blocking the interaction between Fip1, a key component of the cleavage and polyadenylation specificity factor, and the mRNA poly(A) tail[52]. These findings suggest snoRNAs play complex roles in mRNA processing and maturation.
In addition to their direct roles in mRNA processing, snoRNAs also contribute to the regulation of alternative splicing, a more complex layer of gene regulation. Alternative splicing is primarily regulated by RNA-binding proteins that bind to pre-mRNA near different splice sites, yielding alternatively spliced mRNAs[53]. Large-scale snoRNA-RNA interaction models reveal that 30% of snoRNAs interact with host transcripts near alternative splicing exons. Subsequently, by binding proteins that mask the branch point, they inhibit splicing of adjacent exons[54]. This mechanism was validated in SNORD2, which modulates mRNA stability and nonsense-mediated decay by binding intron sequences of its host gene EIF4A2[54]. Furthermore, snoRNAs regulate splicing accuracy by modifying core spliceosome components. For example, SNORD89 was shown to guide 2′-O-methylation at two adjacent sites on U2 snRNA, thereby fine-tuning recognition and selection of splice sites[55]. Moreover, splicing failure similarly leads to hmsnoRNAs production, creating a feedback loop[51]. Additionally, mature snoRNAs can bind to mature mRNAs and directly regulate mRNA stability and translation efficiency. Secreted proteins play crucial roles in cellular communication. The ternary complex formed by SNORA73, mRNAs encoding secreted proteins, and 7SL RNA enhances mRNA binding to signal recognition particles, thereby promoting target protein secretion[56].
RNA derived from snoRNAs
Both H/ACA box and C/D box snoRNAs can be processed into sdRNA. The main difference is that C/D sdRNA does not require Dicer cleavage and is mainly formed from the 5' end of snoRNAs, while H/ACA sdRNA is mainly formed by Dicer cleavage of the 3' end of snoRNAs[57,58]. For example, the U3 snoRNAs are able to be processed into the microRNA miR-U3 by Dicer and Argonaute, however the classical catalytic microRNA component Drosha is not involved in the production of miR-U3[59]. Furthermore, processed snoRNAs (psnoRNA) processed by SNORD115 exhibit a structural deletion of the snoRNA stem, which can target other RNAs and is responsible for heterogeneous nuclear RNA splicing[60]. This greatly broadens the diversity of snoRNAs species and suggests that snoRNAs may be involved in more complex ncRNA regulatory networks.
snoRNAs participate in the DNA damage response
Chromatin-associated RNA (caRNA) is a class of RNA molecules in mammalian cells that associate with chromosomes, playing roles in chromatin loop formation, genomic architecture, and epigenetics[61]. In Drosophila, caRNA primarily consists of snoRNAs that stably bind to chromatin through interaction with the Drosophila chromatin-binding protein Df31, thereby maintaining persistent chromatin accessibility[62]. On the other hand, poly (ADP-ribose) polymerase 1 (PARP1), a widely expressed DNA damage sensor and key catalytic enzyme in eukaryotic nuclei, plays a central role in DNA damage response and repair[63]. Recent studies have revealed complex functional interactions between snoRNAs and PARP1. For example,
Although most of the non-canonical functions of the aforementioned snoRNAs were not directly observed in the cardiovascular system, they exhibit profound underlying associations with cardiovascular pathologies. First, evidence demonstrates that snoRNAs can serve as guide RNAs for NAT10, directing its site-specific modification[48]. Given that NAT10-mediated ac4C modification levels promote myocardial hypertrophy and fibrosis in HF patients and animal models of aortic stenosis[66], this suggests specific snoRNAs may participate in cardiac remodeling by guiding NAT10-mediated ac4C modification. Second, snoRNAs may influence the precise expression of cardiac developmental genes, ion channel proteins, and contraction-related proteins by participating in mRNA alternative splicing and processing. Splicing abnormalities have been established as a pathogenic basis for various cardiomyopathies and arrhythmias[67,68]. Furthermore, given the critical role of sdRNAs in regulating mitochondrial autophagy and aging in chondrocytes, and considering the heart's high dependence on mitochondrial homeostasis as an energy-intensive organ, sdRNAs may exert key protective functions in cardiomyocytes through analogous mitochondrial quality control mechanisms[69]. Finally, snoRNAs play a particularly crucial role in DNA damage responses—myocardial cells undergo substantial oxidative DNA damage during ischemia-reperfusion or stress loading. Combined with their function in regulating key repair molecules like PARP1 in other cell types, snoRNAs may directly influence myocardial cell survival decisions, thereby participating in the pathophysiology of ischemic heart disease. Although definitive evidence revealing the precise mechanisms by which these non-canonical snoRNAs functions regulate CVD development remains elusive, dysfunctional snoRNAs likely contribute extensively to diverse cardiovascular pathologies. This provides novel molecular targets for future cardiovascular disease diagnosis and treatment.
THE ROLE of snoRNAs IN CVD
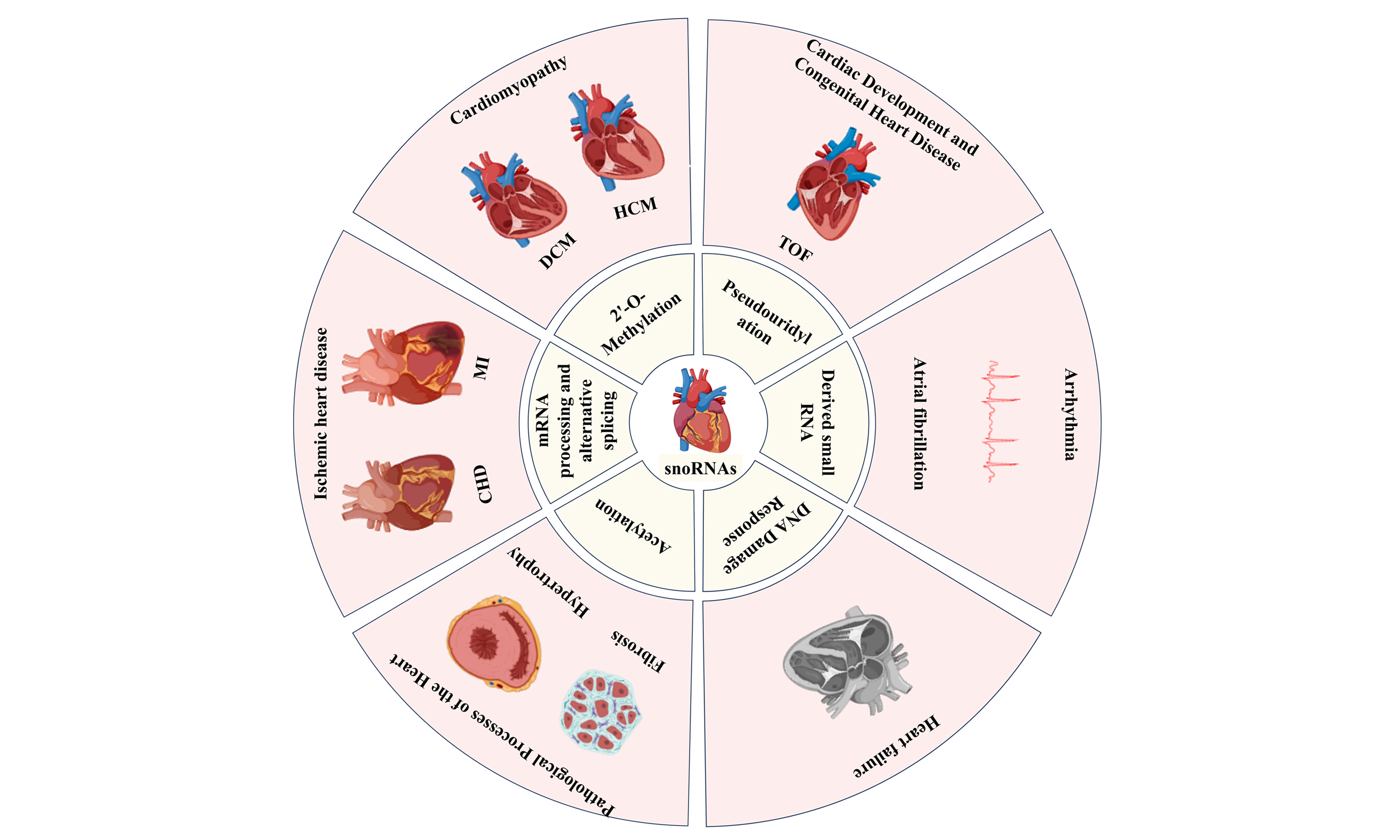
The development of CVD is a complex, multi-stage evolutionary process, with snoRNAs participating throughout its entire progression [Figure 3]. Specifically, this involvement spans from the developmental origins of congenital heart disease, through the pathological changes resulting from adaptive and maladaptive vascular remodeling, to MI caused by coronary heart disease (CHD). Furthermore, snoRNAs play a crucial role in structural and electrical abnormalities inherent to the heart itself, such as primary cardiomyopathies and arrhythmias, ultimately contributing to the onset and progression of end-stage HF in CVD. This chapter will systematically elucidate the specific roles of snoRNAs across various CVD conditions [Table 1].
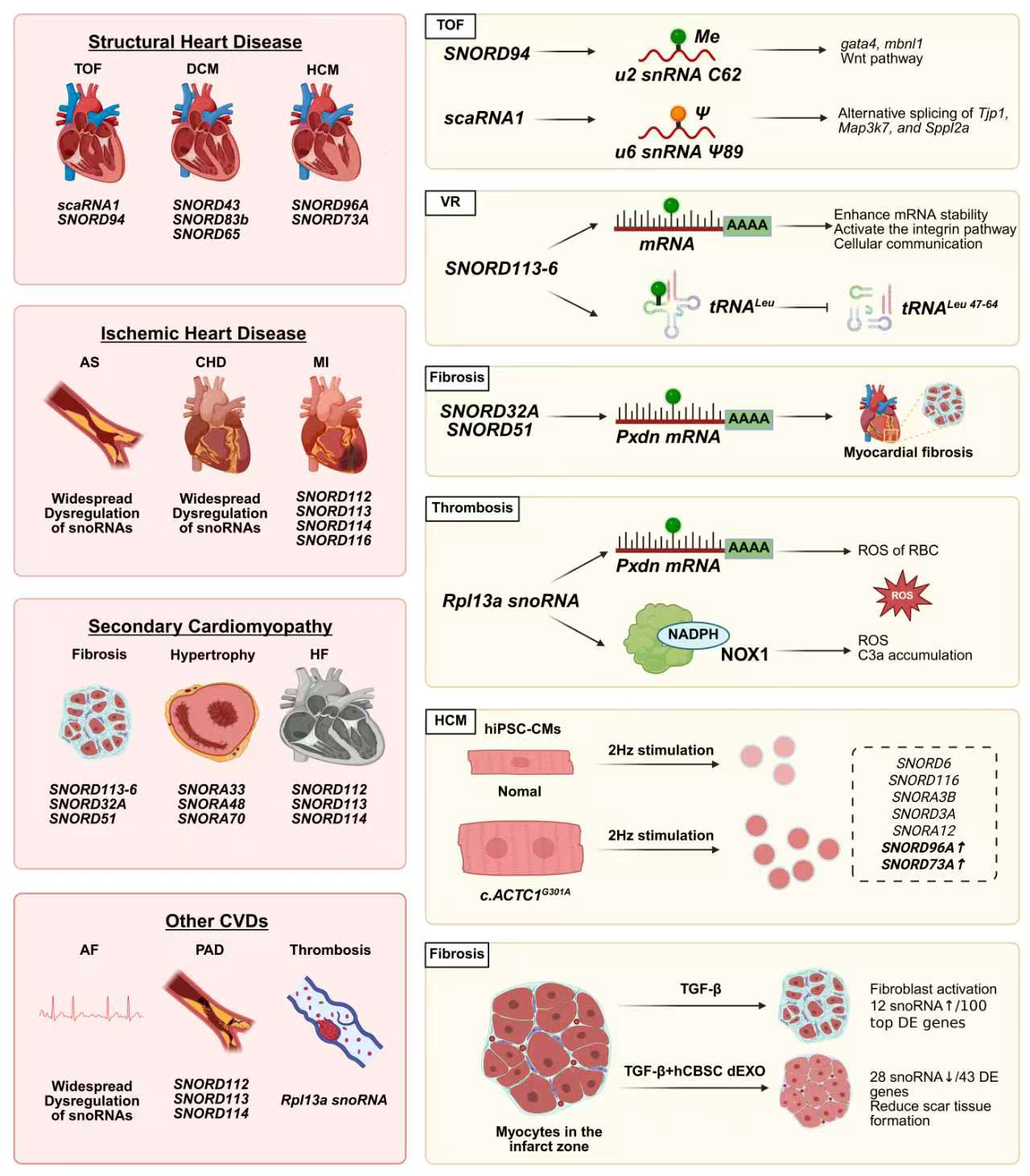
Figure 3. Functions and mechanisms of snoRNAs in cardiovascular diseases. CVD: Cardiovascular disease; TOF: tetralogy of fallot; DCM: dilated cardiomyopathy; HCM: hypertrophic cardiomyopathy; AS: atherosclerosis; CHD: coronary heart disease; MI: myocardial infarction; HF: heart failure; AF: atrial fibrillation; PAD: peripheral artery disease; VR: vascular remodeling; PXDN: peroxidasin; gata4: gata binding protein 4; mbnl1: muscleblind like splicing regulator 1; Tjp1: tight junction protein 1; Map3k7: mitogen-activated protein kinase kinase kinase 7; Sppl2a: signal peptide peptidase like 2A; NOX1: NADPH oxidase 1; RBC: red blood cell; C3a: complement component 3a; hiPSC-CMs: human induced pluripotent stem cell-derived cardiomyocytes; TGF-β: transforming growth factor beta; CBSC-dEXO: cortical bone stem cell-derived exosomes; DE: differential expression; RPL13A: ribosomal protein L13a; tRNALeu: tRNA to protect the leucine anticodon TAA. c.ACTC1: actin alpha cardiac muscle 1. (Created in BioRender. Cao, H. (2026) https://BioRender.com/acu5pfs).
SnoRNAs with functions in CVD
| SnoRNA | Location | Targets/Mechanisms | Expression | Diseases | Species | Model | Refs. |
| Clinical cohort study and clinical specimen analysis | |||||||
| 135 snoRNAs (including scaRNA1 and SNORD94) | - | Downregulated snoRNAs primarily target 4 ncRNAs: rRNA 28S, rRNA 18S, snRNA U2 and snRNA U6 | 126 downregulated, 9 upregulated | TOF | Homo sapiens | Right ventricular myocardium from infants with tetralogy of fallot | [74] |
| 56 snoRNAs (including SNORA15) | - | ND | DE | AS | Homo sapiens | Genome-wide DNA methylation data from the Framingham Heart Study AS Risk Cohort (n = 222) | [97] |
| SNORD114-1 | 14q32 | ND | Upregulated | PAD | Homo sapiens | Comparison of plasma snoRNA expression profiles between patients with end-stage PAD and elite cyclists | [100] |
| SNORD112, SNORD113-2, SNORD113-6, SNORD114-1 | 14q32 | The levels of SNORD113-2 and SNORD114-1 showed a significant negative correlation with platelet activation | Upregulated | PAD | Homo sapiens | Blood samples from patients with end-stage PAD (n = 104) | [16] |
| Rpl13a snoRNA | Rpl13a | 2'-O-methylation of NADPH oxidase 1 mRNA | Upregulated | VT | Homo sapiens | Patients with venous thromboembolism and aged mice with Rpl13a snoRNA knockout | [94] |
| 74 snoRNAs | - | ND | DE | CHD | Homo sapiens | Plasma RNA-seq data from the Framingham Heart Study | [110] |
| 43 snoRNAs | - | ND | DE | CHD | Homo sapiens | Plasma RNA-seq data from the Framingham Heart Study | [111] |
| SNORD112, SNORD113-2, SNORD113-6, SNORD113-7, SNORD113-8, SNORD113-9, SNORD114-1 | 14q32 | ND | Upregulated | ST-segment elevation MI | Homo sapiens | PROspective Study of Pravastatin in the Elderly at Risk(n = 5,244) | [17] |
| 40 snoRNAs | - | ND | 12 upregulated, 28 downregulated | MI | Homo sapiens | NHCFs treated with TGF-β and hCBSC-dEXOs | [118] |
| 12 snoRNAs | - | ND | DE | PCH | Homo sapiens | hiPSC-CMs (c.ACTC1G301A) and isogenic control | [83] |
| SNORD73A, SNORD96A | RPS3A, RACK1 | ND | Upregulated | PCH | Homo sapiens | EVs of hiPSC-CMs (c.ACTC1G301A), 2-Hz stimulation | [83] |
| 31 snoRNA | - | ND | DE | AF | Homo sapiens | A cohort study of patients with valvular heart disease and AF | [119] |
| 14q32 snoRNA | 14q32 | ND | DE | HF | Homo sapiens | PROspective Study of Pravastatin in the Elderly at Risk(n = 5,244) | [17] |
| Animal experiments | |||||||
| SNORD94 | 2p11.2 | 2'-O-Methylation of C62 in U6 snRNA | Downregulated | TOF | Danio rerio | Antisense morpholino knockdown model | [73] |
| scaRNA1 | 1p35.3 | Pseudouridylation modification at nucleotide 89 of U2 snRNA; Regulates the alternative splicing of Tjp1, Map3k7, and Sppl2a | Downregulated | TOF | Coturnix japonica | Antisense morpholino knockdown model | [76] |
| SNORD45A | 1p31.1 | Regulates HIF-1α expression (mRNA and protein levels); promotes endothelial cell migration and angiogenesis | Upregulated | Corneal neovascularization | Rattus norvegicus | Alkali burn-induced corneal neovascularization rat model, cellular pathological model, in vitro endothelial cell model | [87] |
| SNORD32A | Rpl13a | 2′-O-methylation of Pxdn mRNA | Upregulated | CF | Mus musculus | 293T cells, Rpl13a-snoless (-/-) mice | [32] |
| SNORD51 | EEF1B2 | 2′-O-methylation of Pxdn mRNA | - | CF | Mus musculus | 293T cells, Rpl13a-snoless (-/-) mice | [32] |
| 7 snoRNAs (including SNORA33) | - | SNORA33 binds to EZH2 | Upregulated | SCH | Mus musculus | Mice with myocardial hypertrophy at 4 h post-induction with Ang II | [105] |
| SNORA48 | EIF4A1 | ND | Upregulated | SCH | Rattus norvegicus | Spontaneous hypertensive rats with myocardial hypertrophy | [106] |
| SNORA70 | Xq28 | SNORA70 exhibits cardiac tissue specificity | - | SCH | Rattus norvegicus | Rats with hypoxia-induced pulmonary arterial hypertension | [107] |
| Rpl13a snoRNA | Rpl13a | 2′-O-methylation of Pxdn mRNA | Upregulated | VT | Mus musculus | Blood samples from mice across all age groups, as well as from Rpl3a snoRNA conditional knockout mice | [93] |
| MBII-343 b | 12 F1; 12 60.54 cM | MBII-343 is associated with Meg3 | - | UPD14 syndrome | Mus musculus | Distal region of mouse chromosome 12, UPD14 syndrome | [80] |
| SNORD43, SNORD83b, SNORD65 | - | Directs the transcription of ELAC2, thereby guiding 3' tRNA processing | Upregulated | PCH | Mus musculus | ELAC2 conditional knockout mice | [81] |
| 23 snoRNAs | - | ND | Downregulated | PCH | Drosophila melanogaster | Drosophila melanogaster model of feline HCM | [82] |
| Cell experiments | |||||||
| 10 snoRNAs | - | snoR-6253.1 exhibits vascular tissue specificity | 6 upregulated, 4 downregulated | Endotheliitis | Rattus norvegicus | TNF-α stimulation of primary rat endothelial cells | [86] |
| SNORD113-6/ AF357425 | 14q32/DLK1-DIO3 | Play dual role in integrin signaling and arterial fibroblast function via pre-mRNA processing and 2'O-ribose methylation | Upregulated | CF | Mus musculus | Primary mouse fibroblasts | [88] |
| SNORD113-6/AF357425 | 14q32/DLK1-DIO3 | 2'-O-Methylation modifies tRNALeu to protect it from cleavage into tRFLeu 47-64 | Upregulated | CF | Mus musculus Homo sapiens | Hypoxia induction in mouse and human fibroblasts, human arterial tissue | [89] |
The role of snoRNAs in congenital heart disease
Congenital heart disease is a category of birth defects characterized by structural and functional abnormalities of the heart and great vessels arising from genetic and environmental factors during embryonic development. Affected infants exhibit a diverse spectrum of cardiac and great vessel malformations present at birth[70]. For a long time, its etiology was primarily attributed to mutations in coding genes[71]. Emerging evidence indicates that ncRNAs, such as snoRNAs, participate in the fine-tuning regulation of cardiac embryonic development[72,73]. Dysregulation of their expression profiles constitutes an emerging mechanism underlying congenital heart disease development. Reports indicate that over 50% of genes associated with cardiac development exhibit alternative splicing in the right ventricle of infants with Tetralogy of Fallot (TOF)[74]. A comparative study revealed widespread dysregulation of 135 snoRNAs in right ventricular myocardial tissue from TOF patients, with 126 showing significant downregulation. Notably, the snoRNA expression profile in TOF patients exhibited high similarity to fetal heart specimens (n = 115), suggesting their snoRNAs expression pattern remains arrested at the embryonic developmental stage[74]. This evidence strongly indicates snoRNAs play a crucial role in cardiac embryonic development, potentially influencing the expression of key developmental genes and thereby contributing to congenital heart defects. Further studies revealed that 12 scaRNAs participate in abnormal splicing of multiple cardiac developmental genes (including gata4, mbnl1 and Wnt pathway genes) by targeting U2 and U6 snRNAs. In Danio rerio, scaRNA1 or SNORD94 functional deficiencies exhibited altered splicing subtypes of cardiac regulatory genes and cardiac developmental arrest[73]. Mechanistically, SNORD94 deficiency reduces methylation at position C62 of U6 snRNA[75]. In Coturnix japonica, scaRNA1 deficiency decreases Ψ89 levels in U2 snRNA by 5% and guides alternative splicing of Tjp1, Map3k7, and Sppl2a[76]. The RNA modification defects observed in these animal models are consistent with the reduced levels of specific RNA modifications found in the right ventricular myocardium of TOF patients[75,77].
In summary, the abnormal expression of snoRNAs and the splicing dysregulation they mediate offer a novel perspective for understanding the molecular pathomechanisms of congenital heart disease. Notably, the altered expression patterns of snoRNAs in myocardial tissue from patients with Tetralogy of Fallot, coupled with cardiac developmental arrest induced by snoRNA defects in animal models, collectively demonstrate that snoRNA homeostasis is an essential prerequisite for precise embryonic heart development. This evidence reveals a direct link between abnormal snoRNA regulation and cardiac structural malformations. Although advances in prenatal imaging have significantly reduced the birth rate of children with Tetralogy of Fallot, diagnostic accuracy remains limited by fetal cardiac volume, making it challenging to detect potential abnormalities at earlier gestational weeks[78]. Therefore, snoRNAs exhibiting abnormal expression during early embryonic development hold promise as earlier molecular diagnostic markers and potential targets for early intervention, thereby improving postnatal cardiac function in children with Tetralogy of Fallot.
The role of snoRNAs in cardiomyopathy
In addition to participating in the formation of congenital cardiac structural abnormalities during embryonic development, abnormal regulation of snoRNAs also extends to postnatal myocardial structural remodeling. Primary cardiomyopathy is a group of diseases characterized by structural and functional damage to the heart muscle, caused by both genetic and environmental factors. It primarily includes dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM)[79]. Early studies revealed that MBII-343 on mouse chromosome 12 is closely linked to the distal imprinted gene Meg3, whose paternal expression induces severe cardiomyopathy[80]. Among these, the association between DCM and snoRNAs was first elucidated. Dual-targeted ElaC domain protein 2 (ELAC2) is essential for processing nuclear 3'tRNAs. Its processing defect causes combined defects in nuclear and mitochondrial gene expression, ultimately leading to DCM and premature death within 4 weeks[81]. Specifically, ELAC2 deficiency leads to loss of 3'-end tRNA processing function, triggering compensatory accumulation of C/D box snoRNAs. This is particularly evident in the abnormal enrichment of snoRNAs involved in ELAC2 translation (SNORD43, SNORD83b, SNORD65). This indicates ELAC2 functions as a downstream component of snoRNA-mediated RNA modification. In recent years, the potential role of snoRNAs in HCM has garnered significant attention. For instance, in D. melanogaster models of HCM harboring myosin-binding protein C mutant alleles, researchers observed substantial downregulation of 23 snoRNAs in the R820W mutant strain, coinciding with the emergence of a myocardial hypertrophic phenotype[82]. However, the open tubular heart of Drosophila differs significantly from mammalian cardiac structures, and this disparity may render snoRNA targets identified in Drosophila ineffective in mammals. Further studies using human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) revealed 12 differentially expressed snoRNAs, including SNORD6, SNORD116, and SNORA3B, by comparing c.ACTC1G301A mutant and wild-type cohorts. Notably, under high workload conditions (2 Hz), the snoRNA expression profiles of extracellular vesicles (EVs) from these cells exhibited intergroup differences, including SNORD3A and SNORA12. Specifically, compared to EVs from wild-type hiPSC-CMs stimulated at 2 Hz, EVs from HCM hiPSC-CMs stimulated at 2 Hz exhibited significantly increased levels of SNORD96A and SNORD73A[83]. Although this study provides preliminary scientific evidence for snoRNA regulation of HCM development based on human-derived in vitro experiments, relying solely on a single-cell culture system of hiPSC-CMs overlooks the impact of intercellular interactions on snoRNA regulation.
Based on the above evidence, the roles of snoRNAs in congenital heart disease and cardiomyopathy collectively reveal their persistent function in maintaining cardiac development and structural homeostasis. They not only participate in embryonic cardiac morphogenesis but also mediate postnatal adaptation of cardiomyocytes to genetic mutations and mechanical stress. Notably, the expression profiles of snoRNAs in EVs from HCM patients dynamically change with workload alterations. This expands the research scope of snoRNAs beyond traditional ribosomal biosynthesis to the domains of myocardial force sensing and structural remodeling, suggesting that snoRNAs not only function as intracellular regulatory molecules in disease development but may also serve as dynamic biomarkers reflecting myocardial mechanical stress. Furthermore, the detectability of snoRNAs in EVs provides practical evidence for developing snoRNA-based liquid biopsy assays, which may advance precision diagnosis and treatment of cardiovascular diseases.
The role of snoRNAs in vascular remodeling
Moreover, the regulation of cardiac structural integrity by snoRNAs extends beyond maintaining the homeostasis of cardiomyocytes themselves. It further encompasses endothelial cells, smooth muscle cells, and fibroblasts that constitute the vascular wall, profoundly influencing the process of vascular structural remodeling. Vascular remodeling encompasses both adaptive and maladaptive changes in the vascular wall, evolving toward either angiogenesis or the formation of atherosclerosis and aneurysms[84]. In fact, maladaptive vascular remodeling is a key pathway in the pathogenesis of most CVD, particularly involving persistent inflammatory stimulation of endothelial cells, phenotypic switching of vascular smooth muscle cells, and excessive activation of vascular fibroblasts[84,85]. Generally, endothelial cell injury marks the initial stage of vascular damage. Early studies in TNF-α-induced endothelial inflammation revealed widespread dysregulation of snoRNAs, with snoR-6253.1 exhibiting tissue-specific expression in blood vessels[86]. Recent studies on SNORD45A provide compelling evidence supporting the critical role of snoRNAs in vascular endothelial injury and repair[87]. Specifically, SNORD45A promotes corneal endothelial cell proliferation and migration by inducing hypoxia-inducible factor-1α expression, ultimately improving the hypoxic microenvironment of the cornea through participation in angiogenesis. However, endothelial cell-driven angiogenesis is not an isolated event; its outcomes depend on the synergistic actions of vascular smooth muscle cells and vascular fibroblasts. Within vascular fibroblasts, SNORD113-6 was found to play a complex and critical role, with its activation closely associated with the development of myocardial fibrosis. On one hand, SNORD113-6 targets mRNA associated with the integrin pathway, enhancing mRNA stability through 2'-O-methylation modification. This not only promotes the proliferation and migration of vascular fibroblasts themselves but also further regulates local intercellular communication functions within the vasculature, resulting in accelerated vascular wound healing efficiency at the macroscopic level[88]. On the other hand, SNORD113-6 induces 2'-O-methylation of tRNA to protect the leucine anticodon TAA (tRNALeu) from site-specific cleavage during vascular remodeling, thereby preventing the generation of the tRFLeu 47-64 fragment[89]. However, when interpreting the aforementioned synergistic functions of SNORD113-6, its cellular basis must be carefully considered. Notably, both studies relied on the NIH-3T3 fibroblast cell line. While NIH-3T3 serves as a classic model for studying fibroblast biological behavior, it inherently represents a heterogeneous population rather than a clonal population of a single functional subtype[90]. This implies that the SNORD113-6-mediated effects on proliferation, migration, and methylation observed in these studies represent an average of responses across multiple cellular subpopulations. However, with advances in single-cell sequencing and spatial transcriptomics, we now recognize that different fibroblast subtypes exhibit distinct transcriptomes and epigenomes[91,92]. Therefore, there is an urgent need to precisely partition fibroblast subpopulations and utilize single-cell and spatial transcriptomics technologies to pinpoint the functional roles of SNORD113-6 within specific fibroblast subtypes.
Two additional snoRNAs, SNORD32A and SNORD51, participate in translational regulation of fibrosis-associated mRNAs by directing 2′-O methylation of peroxidasin (PXDN) mRNA[32]. SNORD32A localizes to ribosomal protein L13a (Rpl13a). The complex relationship between Rpl13a snoRNAs and venous thrombosis has recently been elucidated. By collecting blood samples from mice of different ages, it was observed that Rpl13a snoRNAs enhance reactive oxygen species production within red blood cells by regulating 2′-O methylation of PXDN mRNA, thereby promoting thrombus size, thrombus red blood cell content, and activation of prothrombin in red blood cells[93]. Furthermore, Rpl13a snoRNAs knockout rats exhibited inhibition of NADPH oxidase 1 (NOX1), which reduced C3a deposition and pre-activation of coagulation during aging[94]. However, mature red blood cells lack the capacity for de novo RNA transcription and processing. The regulation of red blood cells by Rpl13a snoRNAs likely occurs at earlier stages, such as erythroid progenitor cells or reticulocytes. Therefore, the observed phenotypic alterations in red blood cells may not reflect immediate regulation but rather developmental control and aging processes established at the origin of red blood cells.
Overall, C/D box snoRNAs establish a complex molecular regulatory network within the multicellular cooperative mechanisms of the vascular wall: by directing specific RNA 2'-O-methylation modifications, they regulate endothelial cell hypoxia response and proliferation, activate fibroblasts and facilitate intercellular communication, and participate in erythrocyte oxidative stress and thrombosis. These findings suggest that the regulatory network centered on C/D box snoRNAs may hold significant translational value in guiding vascular repair and preventing pathological remodeling. Unfortunately, while the role of C/D box snoRNAs in vascular remodeling has begun to emerge, whether H/ACA box snoRNAs participate in regulating similar vascular pathological processes by directing pseudouridylation remains an unexplored field awaiting further investigation.
The role of snoRNAs in peripheral angiopathic disease
The maladaptive outcome of vascular remodeling ultimately manifests as atherosclerosis (AS). AS is a chronic inflammatory disease affecting large and medium-sized arteries, characterized by lipid deposition in the arterial intima and plaque formation driven by inflammatory responses. It serves as the primary pathological basis for myocardial infarction and stroke[95]. Smoking is an independent risk factor for AS development, exhibiting a robust dose-response relationship[96]. An analysis of whole-blood DNA methylation from an AS risk cohort (n = 222) in the Framingham Health Study revealed that over 50% of the differentially methylated regions in smokers were located within snoRNA genes or their host genes, with notably reduced methylation of SNORA15 compared to non-smokers and ex-smokers[97]. This indicates snoRNAs are a critical link in smoking's impact on the AS epigenetic landscape. When AS primarily affects lower-limb arteries, causing stenosis or occlusion, it is clinically termed peripheral artery disease (PAD)[98]. Clinical manifestations of PAD primarily include leg pain, limited mobility, tissue loss, and major adverse events such as amputation and death[99]. Håkansson et al. reported that by comparing plasma snoRNA expression profiles between end-stage PAD patients and elite cyclists, SNORD114-1 was highly expressed in the PAD cohort and correlated with recovery time from endurance exercise[100]. To further elucidate the underlying mechanisms of atherosclerotic thrombotic events in PAD and the efficacy of surgical interventions, Nossent et al. identified four highly expressed snoRNAs (SNORD112, SNORD113-2, SNORD113-6, and SNORD114-1) in the blood of end-stage PAD patients (n = 104), with SNORD113-2 and SNORD114-1 levels showing significant negative correlations with platelet activation[16]. Notably, platelet activation-driven thrombosis and sustained inflammation play key roles in long-term outcomes of PAD and CVD[101]. It is noteworthy that in healthy individuals, platelets themselves do not express SNORD113-2 and SNORD114-1[100]. So where exactly do these snoRNAs in PAD patients' blood originate? Given that platelets, like red blood cells, lack de novo RNA synthesis and processing, this pathological expression of snoRNAs likely stems from megakaryocytes—the cells that produce platelets—thereby presetting the activation threshold for platelets. Concurrently, the roles of the vascular wall and fibroblasts cannot be overlooked[17,27]. Future studies should further clarify the origin of these snoRNAs and explore their potential value in PAD thrombosis risk stratification and intervention.
Vascular hypertensive diseases, strongly correlated with CVD, represent a leading cause of global mortality, primarily encompassing hypertension and pulmonary arterial hypertension[102]. Common clinical manifestations include fatigue, headaches, and dyspnea. These conditions frequently involve myocardial hypertrophy, with electrocardiograms typically revealing axis deviation. Previous studies indicate that Enhancer of zeste 2 (EZH2)-mediated suppression of Six1 in differentiating cardiac progenitor cells is crucial for maintaining stable gene expression and homeostasis in the postnatal heart, while EZH2 inactivation leads to myocardial hypertrophy[103,104]. Recent studies have revealed a novel EZH2-mediated pathway linked to cardiac hypertrophy: in hypertrophic mice 4 h after angiotensin II induction, EZH2 binding to SNORA33 transiently increases and recovers 24 h post-induction[105]. Furthermore, SNORA7A, SNORA31, SNORA47, SNORA68, SNORD15A, and SNORD104 also show affinity for EZH2 binding[105]. This suggests EZH2 may interact with specific snoRNAs to regulate early pathological mechanisms of hypertrophy. Indeed, ribosomal dysfunction is closely linked to myocardial hypertrophy. Compelling evidence indicates that SNORA48, the most abundant snoRNA in normal rat hearts, is hosted within an intron of the gene encoding the translation initiation factor eukaryotic translation initiation factor 4A1. In contrast, SNORA48 expression is significantly downregulated in hypertensive-induced myocardial hypertrophy models. Mechanistically, SNORA48 dysregulation coincides with ribosomal biogenesis defects induced by Chromosome 3p translational efficiency quantitative trait locus and length-dependent alterations in translation efficiency, linking it to translational abnormalities associated with myocardial hypertrophy[106]. Notably, snoRNA functions may vary across different vascular hypertensive diseases. Comparative analysis of normal rats versus rats with 21-day chronic hypoxia-induced pulmonary hypertension demonstrated no significant differences in cardiac tissue expression of snoRNAs. Specifically, although SNORA70 showed no significant changes in pulmonary hypertension, it exhibited distinct tissue specificity in the heart[107].
Overall, snoRNAs have demonstrated significant potential as epigenetic regulators and circulating biomarkers in the fields of AS and PAD. Future research should focus on the molecular regulatory networks of specific snoRNAs in platelet activation and thrombosis, while exploring their clinical translational value in risk stratification and exercise rehabilitation efficacy assessment for PAD patients. Regarding secondary myocardial hypertrophy, existing evidence highlights the etiological heterogeneity and temporal dynamics of snoRNA regulation: in hypertensive models, snoRNAs exhibit dynamic changes in response to pressure load and participate in early pathological processes by regulating ribosomal function, underscoring the urgent need for refined time-gradient studies to capture their transient effects. In contrast, no significant alterations in cardiac snoRNAs were observed in pulmonary arterial hypertension models. This discrepancy may stem from fundamental differences in left- and right-ventricular loading and molecular adaptive mechanisms between the two diseases. It also necessitates consideration of potential limitations in existing studies regarding sampling timepoints and tissue-specific analysis—specifically, the absence of targeted detection during the critical time window for right ventricular hypertrophy. Therefore, subsequent studies must establish experimental systems with precise temporal gradients to systematically elucidate the precise role of snoRNAs in right ventricular remodeling induced by pulmonary arterial hypertension.
The role of snoRNAs in CHD
Atherosclerosis is the primary cause of coronary heart disease, which ranks among the leading causes of death worldwide[108]. Indeed, substantial evidence indicates differential expression of numerous snoRNAs in CHD. Early studies observed that during cardiac arrest-reperfusion in patients undergoing surgery for chronic ischemic heart disease, silver staining intensity in nucleolar organizer regions exhibited dynamic fluctuations—first rapidly declining and then rebounding. Researchers speculated this process might be associated with changes in rRNA modification functions mediated by snoRNAs[109]. This suggests that snoRNA expression in CHD may not follow a simple activation or suppression pattern but rather a complex and dynamic temporal expression pattern. Therefore, prospective longitudinal studies are urgently needed to confirm the dynamic expression patterns of snoRNAs during CHD progression through time-series monitoring. A study using plasma RNA-seq data from the Framingham Heart Study (FHS) has established a foundational resource by identifying 74 snoRNAs present in human blood, with over 50% (56 out of 74) reproducibly detected in an independent validation cohort[110]. Building on this, an independent study using the same dataset conducted functional association validation. Researchers first confirmed the presence of 74 plasma snoRNAs, then analyzed the 43 most abundant via quantitative reverse transcription polymerase chain reaction (qRT-PCR). However, they ultimately found no association between plasma levels of these snoRNAs and the risk of CHD (n = 286) or stroke (n = 63)[111]. This may be attributed to the relatively limited size of observational cohorts, which may be insufficient to detect effects of smaller magnitude in snoRNAs. Additionally, snoRNAs in CHD may exhibit dynamic evolution patterns similar to those seen in myocardial hypertrophy. Current cross-sectional study designs may average out the temporal effects of snoRNAs and inter-individual variability, thereby obscuring the true association between snoRNAs and CHD. Furthermore, the biological functions of circulating snoRNAs and their mechanisms as long-range molecular mediators remain unclear. True CHD-related snoRNAs may occur exclusively within localized macrophages, vascular fibroblasts, and endothelial cells within the plaque. These genuine signals may be diluted by contaminants in the bloodstream. The true value of these signals requires further clarification through more precise techniques such as spatial transcriptomics.
Based on current evidence, research on snoRNAs in CHD urgently requires shifting from simple differential expression analysis to in-depth exploration of dynamic temporal regulation patterns. Therefore, future studies should expand the scope of transcriptomic screening, establish large-scale prospective cohorts with fine-grained temporal gradient divisions, and thoroughly investigate the molecular mechanisms of specific snoRNAs in coronary atherosclerotic plaque stability and ischemic events.
The role of snoRNAs in MI
As a severe terminal event in the chronic progression of CHD, MI marks the pathological transition from stable plaque accumulation to acute thrombosis[112]. The classic clinical presentation of MI is a widespread crushing pain radiating from the jaw to the abdomen. Initial diagnosis primarily relies on interpreting characteristic electrocardiogram changes, with further classification into ST-segment elevation myocardial infarction and non-ST-segment elevation myocardial infarction based on the presence or absence of ST-segment elevation[113]. Although numerous studies have revealed the regulatory role of snoRNA host genes in myocardial infarction, the direct function and mechanisms of snoRNAs themselves in MI development remain incompletely elucidated[114,115]. Previous reports indicate that microRNAs located on human chromosome 14q32 are highly implicated in CVD pathogenesis[85]. Recently, Håkansson et al. demonstrated enriched expression of a snoRNA cluster on chromosome 14q32 in plasma samples from ST-segment elevation MI patients, comprising seven snoRNAs including SNORD112, SNORD113-2, and SNORD114-1. Among these, plasma expression of SNORD113-2 was upregulated twofold between days 4 and 30 of hospitalization[17]. Therapeutically, cortical bone stem cells (CBSCs) effectively reduce post-MI scar formation and improve cardiac function by modulating macrophage polarization and fibroblast function[116,117]. Further studies indicate that injecting CBSC-derived exosomes into the MI region of mice mitigates ischemia-reperfusion injury in the MI area[118]. Additionally, they found that TGF-β combined with human CBSC-derived exosome treatment significantly suppressed the activation of normal human ventricular fibroblasts, with extensive downregulation of snoRNAs observed in the RNA transcriptome[118]. This may be related to ribosomal instability and altered protein translation induced by snoRNAs.
Overall, these snoRNAs also undergo dynamic evolution during MI, revealing expression fluctuations of the chromosome 14q32 snoRNA cluster within a specific post-infarction time window and their potential involvement in stem cell therapy-mediated myocardial repair. These findings suggest snoRNAs could serve as novel circulating biomarkers for dynamically monitoring MI. By establishing large prospective cohorts to analyze temporal expression patterns of snoRNAs in the blood of myocardial infarction patients, future studies may develop models linking snoRNA expression profiles to infarct staging, ventricular remodeling, and prognosis, thereby advancing snoRNA-guided precision diagnosis and treatment. However, further research is needed at this stage to validate the hypothesis of dynamic snoRNA regulation.
The role of snoRNAs in arrhythmia
Beyond regulating cardiac structural remodeling and hemodynamic dysregulation, snoRNAs also play a crucial role in maintaining cardiac electrophysiological homeostasis, with their dysregulation being closely associated with the onset and progression of arrhythmias. A study on patients with valvular heart disease and AF revealed extensive dysregulation of 51 snoRNAs through Protein ANnotation Diagram ORiented Analysis (PANDORA) sequencing of atrial appendage tissue, with SNORA44, SNORD55, and SNORA72 showing the most significant alterations. Further qRT-PCR validation confirmed widespread dysregulation in 31 snoRNAs, indicating stable differential expression of certain snoRNAs in AF patients. However, no significant intergroup differences were observed for SNORA44, SNORD55, or SNORA72[119]. The conflicting results among these sequencing methods may be attributed to snoRNAs themselves, which possess shorter nucleotide sequences compared to mRNAs and miRNAs, more complex secondary structures, broader post-translational modifications, and the formation of snoRNPs. These characteristics introduce quantitative and annotation biases in conventional qRT-PCR, leading to false negative or false positive outcomes. Consequently, existing omics data may fail to accurately reflect the true expression patterns of snoRNAs within cardiac tissues. Nevertheless, this study represents a pioneering investigation into the expression landscape of snoRNAs in atrial fibrillation, thereby laying the groundwork for elucidating the underlying molecular mechanisms.
The role of snoRNAs in HF
HF represents the terminal stage of progression for all aforementioned cardiac pathologies and serves as the common endpoint where numerous cardiac pathologies converge. HF is a clinical syndrome caused by cardiac abnormalities, confirmed by elevated natriuretic peptide levels or objective evidence of cardiogenic congestion[120]. Håkansson et al. provided compelling evidence for the dysregulation of snoRNAs in HF[17]. They observed that, compared to normal saphenous vein tissue and failed coronary artery bypass grafts, human end-stage HF samples exhibited a marked upregulation of snoRNAs in the DLK1-DIO3 imprinted region of the 14q32 gene cluster[17]. This study indicates that 14q32 snoRNAs play a unique role in HF, and their underlying molecular mechanisms remain to be elucidated.
OPPORTUNITIES AND CHALLENGES IN THE CLINICAL TRANSLATION OF snoRNAs
As described above, snoRNAs demonstrate diagnostic and prognostic value in the pathological processes of various CVDs. First, snoRNAs exhibit excellent stability in bodily fluids, a key advantage for their use as liquid biopsy biomarkers. While current research on snoRNAs as bodily fluid markers has primarily focused on the oncology field, these studies fully reveal the universality of snoRNAs in clinical translation, providing important reference for CVD research. Studies have demonstrated that SNORD16, SNORA73B, SCARNA4, and SNORD49B are significantly elevated in the plasma of breast cancer patients and can be detected even in early-stage cases[121]. Furthermore, incorporating SNORD99 and SNORA50C levels in urinary EVs into a model encompassing multiple genes and obesity/hypertension risk factors elevated the integrated model's accuracy for identifying renal clear cell carcinoma to 0.811 (P = 0.0091)[122]. However, when considering snoRNAs as diagnostic biomarkers for congenital heart disease, a critical safety-related challenge must be addressed: all current expression data are derived from cardiac muscle tissue. If applied to prenatal diagnosis, this would necessitate invasive sampling procedures, which carry inherent risks such as miscarriage and infection. Furthermore, key performance metrics such as diagnostic sensitivity and specificity have not yet been validated in any prospective cohort studies.
In addition, realizing this full potential remains hindered by current technical challenges. For instance, in cell, tissue, and liquid biopsy samples, certain snoRNAs exist as “snoRNA-retained transcripts”. These specialized forms typically feature base deletions within RNA sequences located at or very close to the 5' end. This causes short reads and gene-based transcriptomic analysis to overlook such transcripts, resulting in the loss of critical information about certain snoRNAs[123]. Second, snoRNAs may hold unique value in dynamically monitoring disease states[124]. Numerous studies have observed that snoRNA expression levels in CVD are not static but exhibit highly temporal and load-dependent patterns as disease progresses. This evidence suggests snoRNAs could serve as dynamic biomarkers for CVD and indicators of cardiomyocyte functional status in clinical practice. Conversely, the highly temporal nature of snoRNAs poses significant challenges for their detection. Existing studies lack precise capture of sampling time windows, while cross-sectional research tends to average out these temporal effects, obscuring the true association between snoRNAs and CVD. Furthermore, while certain snoRNA-mediated mechanisms inducing CVD have been elucidated in animal models and human cell experiments, their universality in human settings remains to be demonstrated. This is constrained by species differences, cell line heterogeneity, and the inadequate simulation of real human cell-cell communication and mechanical stress in vitro cultures. Future research should employ humanized animal models, organoids, and technologies like single-cell sequencing and spatial transcriptomics to precisely map snoRNA expression in specific cell subpopulations and elucidate their finely tuned regulatory networks during disease progression. Concurrently, large-scale, prospectively designed clinical cohort studies with finely segmented temporal gradients are essential to capture transient snoRNA effects, thereby revealing their precise role in CVD at the macroscopic level. Therapeutically, delivering snoRNAs poses significant challenges. To our knowledge, only one adeno-associated virus (AAV) vector, SPDD-UG, carrying SNORD44 has been used in laboratory settings via intratumoral injection to reverse SNORD44 levels in colorectal cancer[125]. Key challenges include: (1) Complex subcellular localization of snoRNAs. To perform their canonical functions of guiding rRNA methylation or pseudouridylation, snoRNAs must enter the nucleolus. This necessitates delivery systems that not only traverse the plasma membrane but also breach the nuclear envelope to achieve efficient nucleolar enrichment—a feat far more demanding than delivering mRNAs or siRNAs that act solely in the cytoplasm; (2) Functional redundancy is prevalent within snoRNA families. Therapeutic effects targeting a single snoRNA may be averaged out by compensatory effects from other snoRNAs, failing to fundamentally halt CVD progression[89]; (3) snoRNA expression varies across cardiovascular cell subpopulations. Designing delivery systems to precisely target specific cell types remains dependent on developing cell surface markers; (4) Biosafety of delivery systems. Unlike oncology, cardiovascular therapy aims to enhance cardiac function rather than eliminate cardiomyocytes. This shift demands stricter biosafety controls to prevent chronic toxicity events. An ideal delivery system must avoid chronic cumulative toxicity, immunogenic reactions, or off-target effects induced by the carrier itself (whether viral vectors, metal nanoparticles, inorganic non-metallic materials, or organic liposomes)[126,127]. Any potential long-term toxicity risk is fatal to cardiac tissue. Furthermore, treating snoRNAs as therapeutic targets for embryonic heart development would pose significant safety challenges. Since snoRNAs exert global splicing regulation by guiding snRNA modification, interfering with individual snoRNAs is highly likely to result in off-target effects affecting multiple genes, thereby disrupting the normal development of other organ systems in the embryo. Currently, there is a complete lack of data regarding delivery strategies, dose-response relationships, and long-term safety for interventions during early human pregnancy.
In summary, despite the unique advantages of snoRNAs in liquid biopsy and biomarker applications, their clinical translation is constrained by several factors: highly temporal expression patterns, detection limitations of existing sequencing technologies, inadequacies in clinical study design, functional redundancy among family members, and challenges in targeting and biosafety of delivery systems. Therefore, accumulating more high-quality mechanistic evidence and translational medical research data remains essential to advance the clinical application of snoRNAs.
DISCUSSION
Currently, medical research is focused on elucidating the role of ncRNAs in CVD. Numerous high-quality studies have demonstrated their functions and mechanisms, identifying potential CVD biomarkers[7,8]. Based on the current evidence, research on snoRNA regulation in CVD is gradually shifting from observational studies to in-depth exploration of molecular mechanisms. The most promising molecular groups for translational applications include the snoRNA clusters within the DLK1-DIO3 imprinted region on chromosome 14q32 (encompassing the SNORD113 and SNORD114 families) and snoRNAs associated with Rpl13a. These play pivotal roles in vascular remodeling, PAD, MI, and HF within CVD. This suggests snoRNAs may harbor undiscovered unified mechanisms of action. Systematic functional knockout and gain-of-function experiments on this molecular family are urgently needed to clarify their critical roles in disease progression.
However, to truly understand the complex mechanisms of snoRNAs in CVD, numerous scientific questions remain to be addressed at this stage. Although numerous novel experimental techniques (e.g., enhanced cross-linking immunoprecipitation (CLIP)[55], Nanopore Sequencing[34], Psoralen analysis of RNA interactions and structures (PARIS)[46,47] and PANDORA sequencing[119]) have been applied to explore unknown snoRNA targets However, the vast majority of dysregulated snoRNAs identified in observational studies remain classified as orphan snoRNAs. Furthermore, these methods still face significant challenges due to throughput bottlenecks and high costs. Therefore, systematically exploring the biological functions of orphan snoRNAs is a top priority for future research. Furthermore, existing animal and cell models cannot accurately simulate functional changes in the context of human diseases, limiting the universality of mechanism studies. Concurrently, no systematic research has yet mapped expression differences of snoRNAs across distinct cell subpopulations in the cardiovascular system. There is an urgent need to identify key cell populations mediating their pathogenic effects using single-cell technologies. Finally, existing models often study the effects of individual snoRNAs in isolation. Conventional in vitro overexpression or knockdown experiments may overlook the complex regulatory networks and competitive binding interactions among ncRNAs, potentially deviating from their functional patterns in real physiological environments.
It is worth noting that computational biology simulations and artificial intelligence have emerged as key tools to overcome the snoRNA challenge. By integrating multi-omics data, AI models can efficiently predict interaction networks between snoRNAs and potential target RNAs, enabling high-confidence target predictions for orphan snoRNAs. For instance, GCLSDA focuses on inferring disease associations with snoRNAs; RMBase v3.0 enables precise prediction of snoRNA-mediated chemical modification sites; snoRNA-tRNA resolves snoRNA-tRNA interactions; snoDB 2.0 systematically catalogs snoRNA-protein interactions; and snoKARR-seq enables genome-wide snoRNA-mRNA interaction prediction[36,56,128-130]. The development of these computational tools not only significantly narrows the scope of traditional experimental validation and reduces experimental costs but also holds promise for constructing snoRNA interaction networks in CVD.
However, it is crucial to recognize that the ultimate value of AI predictions lies in clinical validation. While next-generation sequencing and computational modeling can yield promising data, few studies provide definitive mechanistic evidence or clinical diagnostic support. Therefore, alongside AI-based target prediction, emphasis should be placed on clinical cohort studies, particularly large-scale, multicenter, prospectively designed clinical cohort studies. Existing small-sample cross-sectional studies have limited efficacy in revealing snoRNA's role in CVD progression. Future research must expand sample sizes to capture CVD heterogeneity while designing precise temporal gradients to detect dynamic changes and transient effects of snoRNAs. Additionally, exploring vesicle-encapsulated forms of snoRNAs in bodily fluids like blood and urine may offer novel strategies for early CVD prediction and dynamic monitoring.
In summary, deepening our understanding of snoRNA functional mechanisms in CVD is paramount. Moving forward, as the research loop of “AI prediction + mechanism validation + cohort replication” matures, snoRNAs are poised to transition from laboratory discoveries to clinical translation. They may emerge as novel targets for early CVD warning, molecular subtyping, and precision interventions, offering a fresh strategic perspective in the fight against cardiovascular disease.
DECLARATIONS
Acknowledgment
The graphical abstract was created with BioRender.com (Created in BioRender. Cao, H. (2026) https://BioRender.com/a4gmkyr).
Authors’ contributions
Conceptualization, Supervision, Writing - review & editing, Funding acquisition: Wang J
Conceptualization, Supervision, Writing - review & editing: Wang Y
Writing - original draft, Visualization: Han Y
Writing - original draft: Sui J
Visualization: Cao H
Writing - review & editing: Jiang X, Zhang H
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Grammarly (version 14.1258.0, released 2025-12-02) was used solely for language editing with a free personal account. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This review was supported by the Natural Science Foundation of Shandong Province (ZR2019ZD28).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical Approval and Consent to Participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Stark BA, Decleene NK, Desai EC, et al. Global, regional, and national burden of cardiovascular diseases and risk factors in 204 countries and territories, 1990-2023. J Am Coll Cardiol. 2025;86:2167-243.
2. Goh LH, Chong B, Van Der Lubbe SC, et al. The epidemiology and burden of cardiovascular diseases in countries of the Association of Southeast Asian Nations (ASEAN), 1990-2021: findings from the Global Burden of Disease Study 2021. Lancet Public Health. 2025;10:e467-79.
3. Zendjebil S, Steg PG. PCSK9 monoclonal antibodies have come a long way. Curr Atheroscler Rep. 2024;26:721-32.
4. Wang R, Maron BA, Loscalzo J. Multiomics network medicine approaches to precision medicine and therapeutics in cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2023;43:493-503.
5. Arduini A, Fleming SJ, Xiao L, et al. Transcriptional profile of the rat cardiovascular system at single-cell resolution. Cell Rep. 2025;44:115091.
6. Santovito D, Weber C. Non-canonical features of microRNAs: paradigms emerging from cardiovascular disease. Nat Rev Cardiol. 2022;19:620-38.
7. Lee S. Cardiovascular disease and miRNAs: possible oxidative stress-regulating roles of miRNAs. Antioxidants. 2024;13:656.
8. Xie X, Huang M, Ma S, et al. The role of long non-coding RNAs in cardiovascular diseases: a comprehensive review. Non-Coding RNA Res. 2025;11:158-87.
9. Borchardt EK, Martinez NM, Gilbert WV. Regulation and function of RNA pseudouridylation in human cells. Annu Rev Genet. 2020;54:309-36.
10. Duan C, Abola Y, Zhao J, Wang Y. Small nucleolar RNAs in head and neck squamous cell carcinomas. J Dent Res. 2024;104:5-16.
11. Migeot V, Mary Y, Fafard-Couture E, et al. RNase III cleavage sites spread across splice junctions enforce sequential snoRNA processing. EMBO Rep. 2025;26:4675-90.
12. Kiss-László Z, Henry Y, Bachellerie J, Caizergues-Ferrer M, Kiss T. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell. 1996;85:1077-88.
13. Cavaillé J, Nicoloso M, Bachellerie J. Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature. 1996;383:732-5.
14. Wang Y, Fu M, Zheng Z, Feng J, Zhang C. Small nucleolar RNAs: biological functions and diseases. MedComm. 2025;6:e70257.
15. Lin Z, Chang J, Li X, et al. Association of DNA methylation and transcriptome reveals epigenetic etiology of heart failure. Funct Integr Genomics. 2021;22:89-112.
16. Nossent AY, Ektefaie N, Wojta J, et al. Plasma levels of snoRNAs are associated with platelet activation in patients with peripheral artery disease. Int J Mol Sci. 2019;20:5975.
17. Håkansson KEJ, Goossens EAC, Trompet S, et al. Genetic associations and regulation of expression indicate an independent role for 14q32 snoRNAs in human cardiovascular disease. Cardiovasc Res. 2019;115:1519-32.
18. Boivin V, Faucher-Giguère L, Scott M, Abou-Elela S. The cellular landscape of mid-size noncoding RNA. WIREs RNA. 2019;10:e1530.
20. Huo M, Rai SK, Nakatsu K, Deng Y, Jijiwa M. Subverting the canon: novel cancer-promoting functions and mechanisms for snoRNAs. Int J Mol Sci. 2024;25:2923.
21. Deryusheva S, Gall JG. Small, smaller, smallest: minimal structural requirements for a fully functional box C/D modification guide RNA. Biomolecules. 2019;9:457.
22. Deschamps-Francoeur G, Couture S, Abou-Elela S, Scott MS. The snoGloBe interaction predictor reveals a broad spectrum of C/D snoRNA RNA targets. Nucleic Acids Res. 2022;50:6067-83.
23. Trucks S, Hanspach G, Hengesbach M. Eukaryote specific RNA and protein features facilitate assembly and catalysis of H/ACA snoRNPs. Nucleic Acids Res. 2021;49:4629-42.
24. Caton EA, Kelly EK, Kamalampeta R, Kothe U. Efficient RNA pseudouridylation by eukaryotic H/ACA ribonucleoproteins requires high affinity binding and correct positioning of guide RNA. Nucleic Acids Res. 2018;46:905-16.
25. De Zoysa MD, Wu G, Katz R, Yu Y. Guide-substrate base-pairing requirement for box H/ACA RNA-guided RNA pseudouridylation. RNA. 2018;24:1106-17.
26. Izumikawa K, Nobe Y, Ishikawa H, et al. TDP-43 regulates site-specific 2′-O-methylation of U1 and U2 snRNAs via controlling the Cajal body localization of a subset of C/D scaRNAs. Nucleic Acids Res. 2019;47:2487-505.
27. Jorjani H, Kehr S, Jedlinski DJ, et al. An updated human snoRNAome. Nucleic Acids Res. 2016;44:5068-82.
28. Kishore S, Gruber AR, Jedlinski DJ, Syed AP, Jorjani H, Zavolan M. Insights into snoRNA biogenesis and processing from PAR-CLIP of snoRNA core proteins and small RNA sequencing. Genome Biol. 2013;14:R45.
30. Zhang X, Yin Q, Wang H, et al. Species-specific alternative splicing leads to unique expression of sno-lncRNAs. BMC Genomics. 2014;15:287.
31. Kocher MA, Huang FW, Le E, Good DJ. Snord116 post-transcriptionally increases Nhlh2 mRNA Stability: implications for human Prader-Willi syndrome. Hum Mol Genet. 2021;30:1101-10.
32. Elliott BA, Ho H, Ranganathan SV, et al. Modification of messenger RNA by 2′-O-methylation regulates gene expression in vivo. Nat Commun. 2019;10:3401.
33. Zhang D, Li B, Xu H, et al. Identification of FBLL1 as a neuron-specific RNA 2′-O-methyltransferase mediating neuronal differentiation. Proc Natl Acad Sci USA. 2024;121:e2406961121.
34. Li Y, Yi Y, Gao X, et al. 2′-O-methylation at internal sites on mRNA promotes mRNA stability. Mol Cell. 2024;84:2320-36.e6.
35. Vitali P, Kiss T. Cooperative 2′-O-methylation of the wobble cytidine of human elongator tRNAMet (CAT) by a nucleolar and a Cajal body-specific box C/D RNP. Genes Dev. 2019;33:741-6.
36. Zhang M, Li K, Bai J, et al. A snoRNA-tRNA modification network governs codon-biased cellular states. Proc Natl Acad Sci USA. 2023;120:e2312126120.
37. Nir R, Hoernes TP, Muramatsu H, et al. A systematic dissection of determinants and consequences of snoRNA-guided pseudouridylation of human mRNA. Nucleic Acids Res. 2022;50:4900-16.
38. Pan Y, Kierzek E, Kierzek R, Mathews DH, Yu Y. A Ψ-Ψ codon-anticodon pairing in nonsense suppression and translational recoding. Nat Chem Biol. 2026;22:983-94.
39. King TH, Liu B, Mccully RR, Fournier MJ. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol Cell. 2003;11:425-35.
40. Sabe SA, Feng J, Sellke FW, Abid MR. Mechanisms and clinical implications of endothelium-dependent vasomotor dysfunction in coronary microvasculature. Am J Physiol Heart Circ Physiol. 2022;322:H819-41.
41. Yan Q, Zhou J, Wang Z, et al. NAT10-dependent N4-acetylcytidine modification mediates PAN RNA stability, KSHV reactivation, and IFI16-related inflammasome activation. Nat Commun. 2023;14:6327.
42. Yu J, Jin J, Kwon E, et al. Programmable RNA acetylation with CRISPR-Cas13. Nat Chem Biol. 2026;22:969-82.
43. Sas-Chen A, Thomas JM, Matzov D, et al. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature. 2020;583:638-43.
44. Sharma S, Langhendries J, Watzinger P, Kötter P, Entian K, Lafontaine DL. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 2015;43:2242-58.
45. Sharma S, Yang J, Van Nues R, et al. Specialized box C/D snoRNPs act as antisense guides to target RNA base acetylation. PLoS Genet. 2017;13:e1006804.
46. Dudnakova T, Dunn-Davies H, Peters R, Tollervey D. Mapping targets for small nucleolar RNAs in yeast. Wellcome Open Res. 2018;3:120.
47. Lu Z, Zhang QC, Lee B, et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell. 2016;165:1267-79.
48. Bortolin-Cavaillé M, Quillien A, Thalalla Gamage S, et al. Probing small ribosomal subunit RNA helix 45 acetylation across eukaryotic evolution. Nucleic Acids Res. 2022;50:6284-99.
49. Thalalla Gamage S, Howpay Manage SA, Chu TT, Meier JL. Cytidine acetylation across the tree of life. Acc Chem Res. 2024;57:338-48.
50. Liu Y, Demario S, He K, Gibbs MR, Barr KW, Chanfreau GF. Splicing inactivation generates hybrid mRNA-snoRNA transcripts targeted by cytoplasmic RNA decay. Proc Natl Acad Sci USA. 2022;119:e2202473119.
51. Cihlarova Z, Kubovciak J, Sobol M, et al. BRAT1 links Integrator and defective RNA processing with neurodegeneration. Nat Commun. 2022;13:5026.
52. Huang C, Shi J, Guo Y, et al. A snoRNA modulates mRNA 3′ end processing and regulates the expression of a subset of mRNAs. Nucleic Acids Res. 2017;45:8647-60.
53. Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457-63.
54. Bergeron D, Faucher-Giguère L, Emmerichs A, et al. Intronic small nucleolar RNAs regulate host gene splicing through base pairing with their adjacent intronic sequences. Genome Biol. 2023;24:160.
55. Song Z, Bae B, Schnabl S, et al. Mapping snoRNA-target RNA interactions in an RNA-binding protein-dependent manner with chimeric eCLIP. Genome Biol. 2025;26:39.
56. Liu B, Wu T, Miao BA, et al. snoRNA-facilitated protein secretion revealed by transcriptome-wide snoRNA target identification. Cell. 2025;188:465-83.e22.
57. Taft RJ, Glazov EA, Lassmann T, Hayashizaki Y, Carninci P, Mattick JS. Small RNAs derived from snoRNAs. RNA. 2009;15:1233-40.
58. Langenberger D, Çakir MV, Hoffmann S, Stadler PF. Dicer-processed small RNAs: rules and exceptions. J Exp Zool Pt B. 2012;320:35-46.
59. Lemus-Diaz N, Ferreira RR, Bohnsack KE, Gruber J, Bohnsack MT. The human box C/D snoRNA U3 is a miRNA source and miR-U3 regulates expression of sortin nexin 27. Nucleic Acids Res. 2020;48:8074-89.
60. Kishore S, Khanna A, Zhang Z, et al. The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Hum Mol Genet. 2010;19:1153-64.
61. Calandrelli R, Wen X, Charles Richard JL, et al. Genome-wide analysis of the interplay between chromatin-associated RNA and 3D genome organization in human cells. Nat Commun. 2023;14:6519.
62. Schubert T, Pusch MC, Diermeier S, et al. Df31 protein and snoRNAs maintain accessible higher-order structures of chromatin. Mol Cell. 2012;48:434-44.
63. Lin X, Leung KSK, Wolfe KF, et al. XRCC1 mediates PARP1- and PAR-dependent recruitment of PARP2 to DNA damage sites. Nucleic Acids Res. 2025;53:gkaf086.
64. Kim D, Camacho CV, Nagari A, Malladi VS, Challa S, Kraus WL. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol Cell. 2019;75:1270-85.e14.
65. Han C, Sun L, Luo X, et al. Chromatin-associated orphan snoRNA regulates DNA damage-mediated differentiation via a non-canonical complex. Cell Rep. 2022;38:110421.
66. Shi J, Yang C, Zhang J, et al. NAT10 is involved in cardiac remodeling through ac4C-mediated transcriptomic regulation. Circ Res. 2023;133:989-1002.
67. Li P, Qin D, Chen T, et al. Dysregulated Rbfox2 produces aberrant splicing of CaV1.2 calcium channel in diabetes-induced cardiac hypertrophy. Cardiovasc Diabetol. 2023;22:168.
68. Vad OB, Angeli E, Liss M, et al. Loss of cardiac splicing regulator RBM20 is associated with early-onset atrial fibrillation. JACC Basic Transl Sci. 2024;9:163-80.
69. Deng Z, Li C, Hu S, et al. sdRNA-D43 derived from small nucleolar RNA snoRD43 improves chondrocyte senescence and osteoarthritis progression by negatively regulating PINK1/Parkin-mediated mitophagy pathway via dual-targeting NRF1 and WIPI2. Cell Commun Signal. 2025;23:77.
70. Meng X, Song M, Zhang K, et al. Congenital heart disease: types, pathophysiology, diagnosis, and treatment options. MedComm. 2024;5:e631.
71. Rao E, Annavarapu S. Genetics of congenital heart disease: a narrative review of challenges and strategies in identifying novel genes. Cureus. 2025;17:e91674.
72. Wang H, Chen X, Li Y, et al. Non-coding RNAs in congenital heart disease and placental development: Bridging molecular mechanisms to clinical biomarkers and therapies. Non-coding RNA Res. 2026;16:1-20.
73. Patil P, Kibiryeva N, Uechi T, et al. scaRNAs regulate splicing and vertebrate heart development. Biochim Biophys Acta Mol Basis Dis. 2015;1852:1619-29.
74. O'brien JE, Kibiryeva N, Zhou X, et al. Noncoding RNA expression in myocardium from infants with tetralogy of fallot. Circ Cardiovasc Genet. 2012;5:279-86.
75. Ogren A, Kibiryeva N, Marshall J, O’brien JE, Bittel DC. Snord94 expression level alters methylation at C62 in snRNA U6. PLoS ONE. 2019;14:e0226035.
76. Brown M, Earl B, Filla M, Kibiryeva N, O’brien JE, Bittel DC. scaRNA1 expression levels affect alternative splicing of mRNA. Genes. 2025;16:864.
77. Nagasawa CK, Kibiryeva N, Marshall J, O’brien JE, Bittel DC. scaRNA1 levels alter pseudouridylation in spliceosomal RNA U2 affecting alternative mRNA splicing and embryonic development. Pediatr Cardiol. 2020;41:341-9.
78. Lei W, Wen C, Li H, et al. An interpretable deep learning model for first-trimester fetal cardiac screening. NPJ Digit Med. 2025;9:43.
79. Huang S, Li J, Li Q, et al. Cardiomyopathy: pathogenesis and therapeutic interventions. MedComm. 2024;5:e772.
80. Shimoda M, Morita S, Obata Y, Sotomaru Y, Kono T, Hatada I. Imprinting of a small nucleolar RNA gene on mouse chromosome 12. Genomics. 2002;79:483-6.
81. Siira SJ, Rossetti G, Richman TR, et al. Concerted regulation of mitochondrial and nuclear non-coding RNAs by a dual-targeted RNase Z. EMBO Rep. 2018;19:EMBR201846198.
82. Tallo CA, Duncan LH, Yamamoto AH, et al. Heat shock proteins and small nucleolar RNAs are dysregulated in a Drosophila model for feline hypertrophic cardiomyopathy. G3. 2021;11:jkaa014.
83. James V, Nizamudeen ZA, Lea D, et al. Transcriptomic analysis of cardiomyocyte extracellular vesicles in hypertrophic cardiomyopathy reveals differential snoRNA cargo. Stem Cells Dev. 2021;30:1215-27.
84. Epstein FH, Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330:1431-8.
85. Wang S, Yang L, Zhao A, et al. A novel hidden protein p-414aa encoded by circSETD2(14,15) inhibits vascular remodeling. Circulation. 2025;151:1568-82.
86. Liu P, Hu L, Shi Y, et al. Changes in the small RNA expression in endothelial cells in response to inflammatory stimulation. Oxid Med Cell Longev. 2021;2021:8845520.
87. Yang X, Li M, Wang H, et al. SNORD45A affects content of HIF-1α and promotes endothelial angiogenic function. Appl Biochem Biotechnol. 2024;196:7185-97.
88. Van Ingen E, Van Den Homberg DAL, Van Der Bent ML, et al. C/D box snoRNA SNORD113-6/AF357425 plays a dual role in integrin signalling and arterial fibroblast function via pre-mRNA processing and 2′O-ribose methylation. Hum Mol Genet. 2022;31:1051-66.
89. Van Ingen E, Engbers PA, Woudenberg T, et al. C/D box snoRNA SNORD113-6 guides 2′-O-methylation and protects against site-specific fragmentation of tRNALeu(TAA) in vascular remodeling. Mol Ther Nucleic Acids. 2022;30:162-72.
90. Rahimi AM, Cai M, Hoyer-Fender S. Heterogeneity of the NIH3T3 fibroblast cell line. Cells. 2022;11:2677.
91. Youness M, Ekhteraei-Tousi S, Nagaraju CK, et al. Location and aetiology are determinants of fibroblast activation and heterogeneity in the failing human heart. Genome Med. 2025;17:155.
92. Amrute JM, Luo X, Penna V, et al. Targeting immune-fibroblast cell communication in heart failure. Nature. 2024;635:423-33.
93. Chauhan W, Sj S, Ferdowsi S, Sohel A, Zennadi R. Red blood cell Rpl13a small noncoding nucleolar RNAs guides 2'-O-methylation on peroxidasin messenger RNA promoting venous thrombosis in aging. J Thromb Haemost. 2025:S1538783625001357.
94. Chauhan W, Ferdowsi S, Sudharshan S, Zennadi R. Rpl13a snoRNAs-regulated NADPH oxidase 1-dependent ROS generation: a novel RBC pathway mediating complement C3a deposition and triggering thrombosis in aging and venous blood clotting disorders. Free Radic Biol Med. 2025;230:138-50.
95. Engelen SE, Robinson AJB, Zurke Y, Monaco C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat Rev Cardiol. 2022;19:522-42.
96. Yao Z, Tasdighi E, Dardari ZA, et al. Association between cigarette smoking and subclinical markers of cardiovascular harm. J Am Coll Cardiol. 2025;85:1018-34.
97. Keshawarz A, Joehanes R, Guan W, et al. Longitudinal change in blood DNA epigenetic signature after smoking cessation. Epigenetics. 2021;17:1098-109.
98. Aboyans V, Canonico ME, Chastaingt L, et al. Peripheral artery disease. Nat Rev Dis Primers. 2025;11:68.
99. Golledge J. Update on the pathophysiology and medical treatment of peripheral artery disease. Nat Rev Cardiol. 2022;19:456-74.
100. Håkansson KEJ, Sollie O, Simons KH, Quax PHA, Jensen J, Nossent AY. Circulating small non-coding RNAs as biomarkers for recovery after exhaustive or repetitive exercise. Front Physiol. 2018;9:1136.
101. Jain K, Tyagi T, Gu SX, Faustino EVS, Hwa J. Demographic diversity in platelet function and response to antiplatelet therapy. Trends Pharmacol Sci. 2025;46:78-93.
102. Sayer M, Webb DJ, Dhaun N. Novel pharmacological approaches to lowering blood pressure and managing hypertension. Nat Rev Cardiol. 2025;22:649-63.
103. Delgado-Olguín P, Huang Y, Li X, et al. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012;44:343-7.
104. He A, Ma Q, Cao J, et al. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ Res. 2012;110:406-15.
105. Wang S, Guo N, Li S, et al. EZH2 dynamically associates with non-coding RNAs in mouse hearts after acute angiotensin II treatment. Front Cardiovasc Med. 2021;8:585691.
106. Witte F, Ruiz-Orera J, Mattioli CC, et al. A trans locus causes a ribosomopathy in hypertrophic hearts that affects mRNA translation in a protein length-dependent fashion. Genome Biol. 2021;22:191.
107. Wang J, Kuang J, Zhang S, et al. Comprehensive characterization of small noncoding RNA profiles in hypoxia-induced pulmonary hypertension (HPH) rat tissues. iScience. 2024;27:108815.
108. Heusch G, Kleinbongard P. The spleen in ischaemic heart disease. Nat Rev Cardiol. 2025;22:497-509.
109. Mamaev NN, Kovalyeva OV, Amineva K, et al. AgNORs in cardiomyocytes from surgical patients with coronary heart disease. Mol Pathol. 1998;51:218-21.
110. Freedman JE, Gerstein M, Mick E, et al. Corrigendum: Diverse human extracellular RNAs are widely detected in human plasma. Nat Commun. 2016;7:11902.
111. Mick E, Shah R, Tanriverdi K, et al. Stroke and circulating extracellular RNAs. Stroke. 2017;48:828-34.
112. Chapman AR, Taggart C, Boeddinghaus J, Mills NL, Fox KAA. Type 2 myocardial infarction: challenges in diagnosis and treatment. Eur Heart J. 2025;46:504-17.
113. Pan W, An Y, Guan Y, Wang J. MCA-net: a multi-task channel attention network for Myocardial infarction detection and location using 12-lead ECGs. Comput Biol Med. 2022;150:106199.
114. Shumliakivska M, Fischer A, Muhly-Reinholz M, et al. The long non-coding RNA Snhg15 protects the heart after myocardial infarction. J Mol Cell Cardiol. 2026;210:72-82.
115. Wang S, Cheng Z, Chen X, Lu G, Zhu X, Xu G. Long noncoding RNA SNHG4 attenuates the injury of myocardial infarction via regulating miR-148b-3p/DUSP1 axis. Cardiovasc Ther. 2022;2022:1-15.
116. Hobby ARH, Berretta RM, Eaton DM, et al. Cortical bone stem cells modify cardiac inflammation after myocardial infarction by inducing a novel macrophage phenotype. Am J Physiol Heart Circ Physiol. 2021;321:H684-701.
117. Yang Y, Johnson J, Troupes CD, et al. miR-182/183-Rasa1 axis induced macrophage polarization and redox regulation promotes repair after ischemic cardiac injury. Redox Biol. 2023;67:102909.
118. Schena GJ, Murray EK, Hildebrand AN, et al. Cortical bone stem cell-derived exosomes’ therapeutic effect on myocardial ischemia-reperfusion and cardiac remodeling. Am J Physiol Heart Circ Physiol. 2021;321:H1014-29.
119. Zeng Y, Yuan Z, Li J, et al. Small non-coding RNA signatures in atrial appendages of patients with atrial fibrillation. J Cell Mol Med. 2024;28:e18483.
120. Bauersachs J, De Boer RA, Lindenfeld J, Bozkurt B. The year in cardiovascular medicine 2021: heart failure and cardiomyopathies. Eur Heart J. 2022;43:367-76.
121. Li X, Zhao X, Xie L, Song X, Song X. Identification of four snoRNAs (SNORD16, SNORA73B, SCARNA4, and SNORD49B) as novel non-invasive biomarkers for diagnosis of breast cancer. Cancer Cell Int. 2024;24:55.
122. Grützmann K, Salomo K, Krüger A, et al. Identification of novel snoRNA-based biomarkers for clear cell renal cell carcinoma from urine-derived extracellular vesicles. Biol Direct. 2024;19:38.
123. Rambaldelli G, Asghar S, Venturi G, et al. Characterization of small nucleolar RNA retaining transcripts in human normal and cancer cells. Non-Coding RNA Res. 2025;13:153-61.
124. Chabronova A, Holmes TL, Hoang DM, et al. SnoRNAs in cardiovascular development, function, and disease. Trends Mol Med. 2024;30:562-78.
125. Yuan S, Wu Y, Wang Y, Chen J, Chu L. An oncolytic adenovirus expressing SNORD44 and GAS5 exhibits antitumor effect in colorectal cancer cells. Hum Gene Ther. 2017;28:690-700.
126. Wang W, Sayedahmed EE, Mittal SK. Significance of preexisting vector immunity and activation of innate responses for adenoviral vector-based therapy. Viruses. 2022;14:2727.
127. Wang X, Zhong X, Li J, Liu Z, Cheng L. Inorganic nanomaterials with rapid clearance for biomedical applications. Chem Soc Rev. 2021;50:8669-742.
128. Xuan J, Chen L, Chen Z, et al. RMBase v3.0: decode the landscape, mechanisms and functions of RNA modifications. Nucleic Acids Res. 2024;52:D273-84.
129. Zhang L, Chen M, Hu X, Deng L. Graph convolutional network and contrastive learning small nucleolar RNA (snoRNA) disease associations (GCLSDA): predicting snoRNA-disease associations via graph convolutional network and contrastive learning. Int J Mol Sci. 2023;24:14429.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.