


Patient-derived tumor organoid models for functional precision oncology in hepatocellular carcinoma
 0
0 
Abstract
Hepatocellular carcinoma (HCC) is characterized by marked intertumoral and intratumoral heterogeneity, which contributes to highly variable responses to systemic therapy and limits the clinical utility of current biomarker-driven precision strategies. Growing evidence suggests that static molecular profiling alone cannot fully capture the functional determinants of therapeutic response, particularly for immune checkpoint-based treatment. In this review, we discuss the concept of functional precision oncology in HCC and provide a structured framework for positioning patient-derived tumor models across the translational spectrum. We compare long-term expandable patient-derived organoids (PDOs), short-term patient-derived organotypic tumor spheroids (PDOTS), and patient-derived tumor fragments (PDTFs), emphasizing their distinct biological scopes and translational roles. Whereas conventional PDOs support scalable assessment of tumor cell-intrinsic drug sensitivity, organotypic platforms preserve endogenous immune and stromal components and therefore permit direct ex vivo interrogation of immunotherapy responses. We further discuss how functional drug-response data generated from these models can serve as phenotypic ground truth for interpreting multi-omics data and informing computational and AI-assisted predictive frameworks. Finally, we outline key challenges related to standardization, scalability, and clinical integration, and propose a roadmap for incorporating patient-derived functional tumor models into precision therapy development for HCC.
Keywords
INTRODUCTION
Hepatocellular carcinoma (HCC) remains one of the leading causes of cancer-related mortality worldwide, and both its incidence and mortality have continued to rise over recent decades[1,2]. Because early-stage disease is often clinically silent, many patients are diagnosed only after curative options such as surgical resection or liver transplantation are no longer feasible[3,4]. Systemic therapy has therefore become a cornerstone of treatment for advanced HCC[5].
The therapeutic landscape of advanced HCC has expanded substantially with the introduction of multikinase inhibitors and immune checkpoint inhibitors (ICIs). Agents such as sorafenib and lenvatinib, together with monoclonal antibodies targeting the PD-1 (programmed cell death protein 1)/PD-L1 (programmed death-ligand 1) axis, have improved survival and reshaped standard-of-care strategies[6-8]. Even so, objective response rates remain modest, and outcomes vary widely among patients receiving the same regimen[9,10]. This marked interpatient heterogeneity remains a major barrier to effective precision therapy in HCC[11].
To improve therapeutic stratification, numerous molecular and immunological biomarkers have been proposed, including PD-L1 expression, tumor mutational burden (TMB), genomic alterations, and transcriptomic signatures[12-14]. However, accumulating clinical evidence indicates that most of these markers provide only probabilistic, population-level associations with treatment response and lack robust predictive power for individual patients[14]. For immune checkpoint blockade in particular, no currently available biomarker has shown sufficient sensitivity and specificity to reliably guide clinical decision-making in HCC[15,16].
Response to systemic therapy, especially immunotherapy, arises from dynamic interactions among tumor cells, immune populations, and the surrounding tumor microenvironment[17,18]. HCC develops within a distinctive hepatic milieu shaped by chronic inflammation, cirrhosis-associated fibrosis, and etiologic drivers such as HBV (hepatitis B virus), HCV (hepatitis C virus), and NASH (non-alcoholic steatohepatitis). These liver-specific features influence immune composition and therapeutic responsiveness and should therefore be considered when interpreting functional tumor models. Static molecular features captured at a single time point cannot fully reflect a tumor’s capacity to respond to a defined therapeutic perturbation[19,20]. This limitation has prompted a conceptual shift in precision oncology, from reliance on correlative biomarkers toward functional assays that directly measure drug-induced responses in patient-derived tumor material, an approach increasingly described as functional precision oncology[21,22].
A range of preclinical models has been used to approximate drug response in HCC. Conventional two-dimensional (2D) cancer cell lines are scalable and experimentally convenient, but they fail to capture intratumoral heterogeneity and the complexity of the tumor microenvironment[23]. Patient-derived xenograft (PDX) models preserve aspects of tumor architecture and genetics and have long been regarded as a preclinical reference standard[24,25]. However, their long establishment time, high cost, and lack of a functional human immune system severely limit their utility, particularly for immunotherapy research[21,26].
Against this background, patient-derived organoid (PDO)-based models have emerged as an attractive intermediate between reductionist in vitro systems and in vivo models. PDOs can be generated within clinically relevant timeframes and retain key genetic and histopathological features of the original tumors, enabling parallel drug testing and mechanistic analysis[27]. More recently, advanced organotypic platforms such as patient-derived organotypic tumor spheroids (PDOTS) and patient-derived tumor fragments (PDTFs) have been developed to preserve native immune and stromal components more effectively[28,29]. These systems make it possible to interrogate functional immune responses to therapeutic agents, including immune checkpoint blockade, in an intact tumor ecosystem ex vivo.
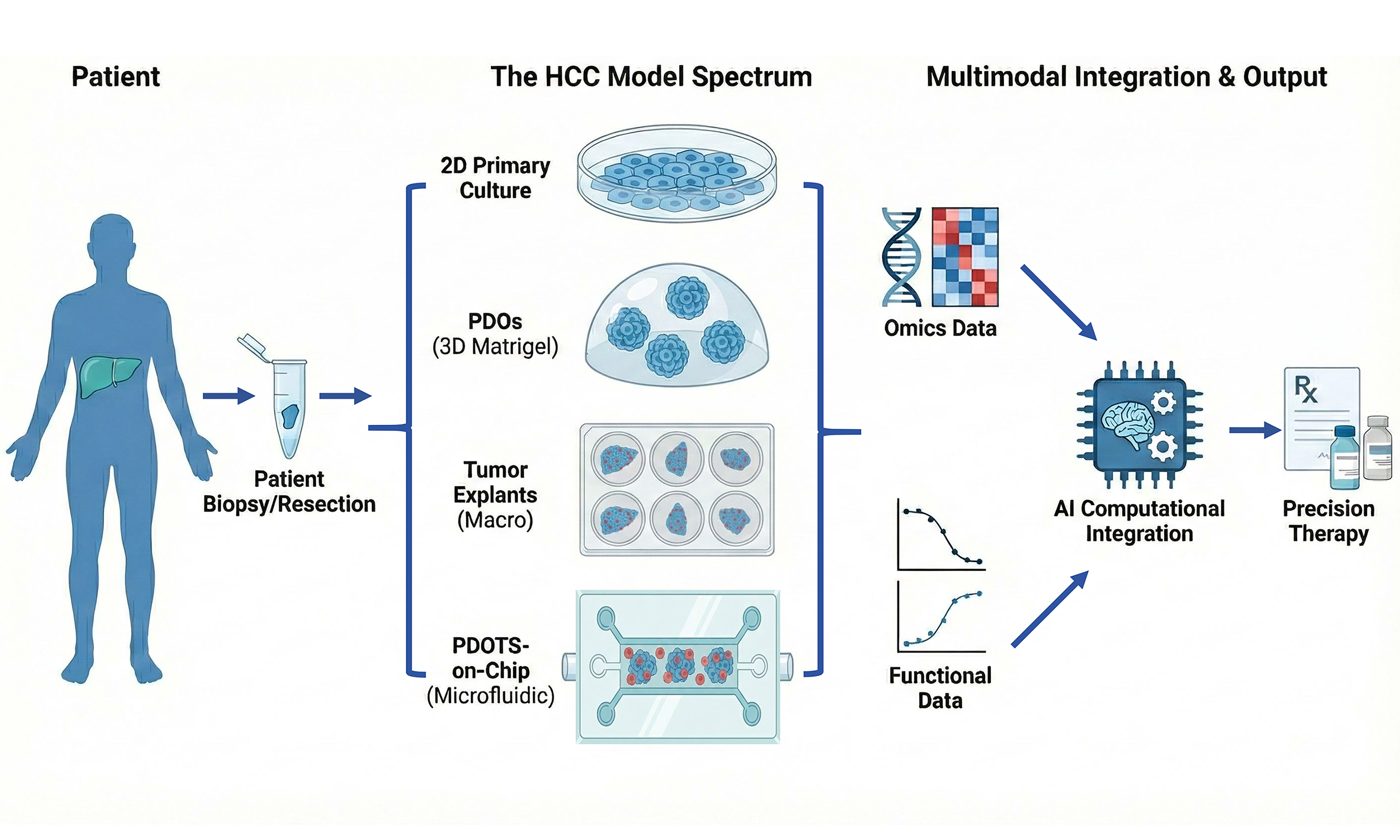
Systematic evaluation of available preclinical HCC models, particularly PDO-based and organotypic platforms, is therefore essential for defining their respective strengths, limitations, and translational value. In this review, we place organoid-based models within the broader landscape of HCC preclinical research, summarize representative applications in drug testing and immunotherapy response assessment, and discuss their emerging role in precision therapy development. Within this framework, functional precision oncology refers to an approach that complements molecular profiling by directly measuring therapy-induced responses in patient-derived tumor models. By integrating functional drug-response assays with genomic and transcriptomic data, this strategy seeks to connect molecular states with experimentally validated therapeutic phenotypes and thereby improve the interpretability and predictive potential of precision oncology.
PRECLINICAL MODELS FOR HCC: A COMPARATIVE LANDSCAPE AND TRANSLATIONAL POSITIONING
Traditional preclinical models in HCC research
2D cell line models: accessibility at the cost of predictive power
2D HCC cell lines are among the earliest and most widely used systems for studying liver cancer biology and drug response. Lines such as HepG2 (human hepatoblastoma-derived cell line), Huh7 and PLC/PRF/5 (human hepatocellular carcinoma cell lines) have been extensively used to interrogate oncogenic signaling, metabolic reprogramming, and sensitivity to cytotoxic or targeted agents[30-32].
However, large pharmacogenomic studies have shown that drug responses observed in 2D cultures often fail to translate into clinical efficacy[33,34]. This limitation largely reflects clonal selection during long-term in vitro propagation, loss of intratumoral heterogeneity, and the absence of three-dimensional architecture and microenvironmental cues[35-37]. In HCC, where stromal interactions and immune contexture strongly influence therapeutic response, these shortcomings are particularly relevant and can produce misleading drug-sensitivity signals[38].
Accordingly, although 2D cell lines remain valuable for mechanistic studies and early-stage compound screening, their ability to predict patient-specific drug response in HCC is inherently limited.
PDX models: in vivo fidelity with limited clinical timeliness
PDX models are generated by engrafting fresh human HCC tissue into immunodeficient mice and have been widely used in preclinical cancer research[39,40]. Multiple studies have shown that HCC PDX models preserve histopathological features, genomic alterations, and interpatient heterogeneity more faithfully than conventional cell line-based systems[41]. They have also been used extensively to evaluate targeted therapies, including sorafenib and lenvatinib, and to investigate mechanisms of drug resistance[42,43]. In addition, patient-specific growth patterns and treatment responses can often be maintained across serial passages, supporting the value of PDX models in late-stage preclinical development[40].
Despite these strengths, several intrinsic limitations restrict the translational utility of PDX models. Establishment usually requires several months, which precludes real-time therapeutic decision-making[44,45]. More importantly, the absence of a functional human immune system fundamentally limits their relevance for immunotherapy research, which is now central to HCC treatment. Although humanized PDX models have been developed, their technical complexity, variable immune reconstitution, and limited standardization have hindered widespread use[46,47].
Genetically engineered mouse models: mechanistic insight without patient specificity
Genetically engineered mouse models (GEMMs) have played a central role in elucidating the molecular drivers of hepatocarcinogenesis[48]. By introducing defined alterations, such as MYC (MYC proto-oncogene, bHLH transcription factor) activation, β-catenin activation, or TP53 (tumor protein p53) loss, GEMMs allow controlled investigation of oncogenic pathways and tumor-immune interactions in an immunocompetent setting[49-51].
Nevertheless, GEMMs are intrinsically limited by species-specific biology and restricted genetic diversity[52]. Tumors arising in these models often follow relatively uniform evolutionary trajectories and do not capture the extensive heterogeneity of human HCC[53]. Thus, while GEMMs are indispensable for mechanistic and hypothesis-driven studies, their ability to predict individual patient responses to systemic therapy remains limited[52].
Emergence of PDO-based models
PDOs: reconstructing tumor cell-intrinsic drug responses
PDOs have emerged as a versatile ex vivo platform that retains key genetic and histopathological features of primary HCC tumors[53,54]. Seminal studies showed that liver cancer organoids preserve driver mutations, copy-number alterations, and lineage-associated transcriptional programs from their parental tumors. This fidelity is substantial but not absolute, because subclonal complexity and some complex structural alterations may be underrepresented or may shift during in vitro propagation as a result of culture-induced selection[27,45].
In functional applications, PDOs have been used successfully for drug-sensitivity testing with cytotoxic and targeted agents[54]. Organoid-based screening in biliary tract cancer and liver cancer cohorts has shown encouraging concordance with patient responses, supporting their utility for predicting chemotherapy efficacy[55,56]. However, because these systems are composed largely of epithelial tumor cells and lack immune and stromal compartments, their value for predicting immunotherapy response is limited.
PDOTS and organotypic tumor spheroids: preserving functional tumor ecosystems
Conventional PDOs are powerful platforms for modeling tumor-intrinsic biology and drug sensitivity, but their epithelial-only nature fundamentally limits their usefulness for immunotherapy research[26,57]. The extensive enzymatic dissociation required for long-term organoid expansion removes endogenous immune and stromal components and therefore precludes direct interrogation of immune-mediated tumor control. Although immune cells can be reintroduced through co-culture approaches, these systems usually rely on peripheral or exogenously added immune cells that lack prior antigen experience within the tumor microenvironment. As a result, they do not faithfully recapitulate patient-specific responses to immune checkpoint blockade[58].
To address these limitations, PDOTS were developed as an alternative culture strategy designed to preserve the native tumor immune ecosystem. PDOTS are generated through minimal dissociation of freshly resected tumor tissue, allowing retention of tumor-resident immune cells within multicellular spheroids. Early landmark studies in melanoma showed that PDOTS could support short-term ex vivo assessment of anti-PD-1 therapy and revealed heterogeneous immune-mediated tumor killing that closely paralleled clinical sensitivity and resistance[29,59]. These studies provided the first functional evidence that immunotherapy responsiveness could be evaluated ex vivo in patient-derived tumor material while endogenous immune context was preserved.
A defining feature of PDOTS is the retention of tumor-infiltrating immune cells that have undergone antigen exposure and immune selection in situ. Unlike peripheral blood-derived or in vitro-expanded immune cells, the populations preserved in PDOTS are tissue-resident and antigen-experienced, having been shaped by sustained interactions with tumor cells, stromal elements, and local immunosuppressive cues[58]. PDOTS therefore capture immune effector states that more closely reflect a patient’s pretreatment immune landscape and provide a biologically relevant substrate for evaluating immune checkpoint blockade.
Consistent with this view, recent high-resolution analyses of patient-derived tumor spheroids and minimally dissociated tumor cultures have shown enrichment of tumor-reactive heterotypic CD8+ T-cell clusters[60]. These clusters contain CD8⁺ T cells physically engaged with autologous tumor cells and/or antigen-presenting cells, forming functional immune synapses that are largely absent from conventional immune co-culture systems. CD8⁺ T cells isolated from such clusters show increased TCR clonality, enrichment of tumor-reactive transcriptional programs, and enhanced cytotoxic capacity, whereas dissociated single T cells more often display bystander or virus-reactive phenotypes with limited antitumor activity[61]. These observations offer a mechanistic explanation for why PDOTS-based platforms outperform simple organoid-immune co-cultures in capturing clinically relevant immune responses.
PDOTS and related organotypic tumor spheroid systems have now been reported in multiple tumor types, including melanoma, non-small-cell lung cancer, and HCC[26,29,58]. Across these settings, they have been used for short-term functional assessment of immune checkpoint blockade, evaluation of immune-mediated tumor killing, and exploration of combination strategies aimed at overcoming primary or acquired immunotherapy resistance. Taken together, these studies establish PDOTS as a reproducible and versatile platform for functional immunotherapy profiling in solid tumors.
PDTFs: capturing early immune reactivation
PDTFs represent an extreme form of organotypic modeling in which intact tissue architecture and spatial immune organization are preserved over short culture periods[62,63]. A landmark study showed that ex vivo PD-1 blockade in PDTFs induced rapid immune reactivation within 24-48 h and that the magnitude of this response correlated closely with clinical outcomes across multiple cancer types[64]. These findings provided direct evidence that functional immune responsiveness can be measured ex vivo in patient-derived tissue and offered a complementary strategy to molecular biomarker-based prediction.
Despite their exceptional biological realism, PDTFs are constrained by practical and methodological limitations that prevent broad translational deployment[63]. The same features that confer their value, namely preserved architecture, spatial immune organization, and minimal perturbation, also restrict culture duration, scalability, and assay repeatability[62]. PDTFs usually remain viable only within a narrow window of approximately 24-72 h, during which immune-cell function progressively declines, limiting analysis largely to early immune activation events[65,66].
In addition, PDTFs require substantial amounts of fresh tumor tissue and do not allow parallel testing of many therapeutic conditions, making them unsuitable for high-throughput screening, longitudinal functional assessment, or iterative regimen optimization[67]. These constraints limit their use as routine predictive tools in clinical decision-making. Instead, their main value lies in serving as high-fidelity proof-of-concept platforms that demonstrate how responsiveness to immune checkpoint blockade is encoded within the pretreatment tumor immune ecosystem and can be revealed ex vivo.
Why organoid-based models complement - rather than replace - traditional systems
Collectively, existing preclinical models capture distinct aspects of HCC biology and treatment response [Table 1]. PDX models are well-suited for long-term in vivo efficacy evaluation, GEMMs provide mechanistic insight in defined genetic settings, and PDOs enable scalable assessment of tumor cell-intrinsic drug sensitivity. Organotypic platforms such as PDOTS and PDTFs bridge these systems by allowing rapid, patient-specific functional assessment while preserving key components of the tumor microenvironment. Rather than replacing traditional models, PDO-based and organotypic systems fill a critical translational gap, especially for immunotherapy, by providing timely and functionally informative readouts that are otherwise difficult to obtain. Among them, organotypic functional platforms are particularly valuable because they address one of the major bottlenecks in contemporary HCC treatment: the inability to prospectively evaluate immune-mediated therapeutic responses at the individual-patient level.
Landscape of preclinical models used in hepatocellular carcinoma research
| Model type | Advantages | Limitations | Typical applications | References |
| 2D primary tumor cell cultures | Rapid establishment; low cost; suitable for short-term functional assays; partially patient-derived | Rapid loss of tumor heterogeneity; absence of 3D structure and tumor microenvironment; high false-positive drug sensitivity | Early-stage drug screening; short-term mechanistic studies | [103-105] |
| PDX | Preserves histopathology and genomic features; reflects interpatient heterogeneity; robust in vivo drug efficacy evaluation | Long establishment time; high cost; lack of functional human immune system; limited suitability for real-time prediction | Preclinical validation of targeted therapies; resistance mechanism studies | [24,40,46] |
| GEMMs | Defined genetic background; immunocompetent environment; strong mechanistic interpretability | Limited genetic diversity; species-specific differences; poor patient-level predictability | Tumorigenesis studies; immune-oncology mechanism research | [52,106,107] |
| PDOs | Retain tumor genetic and histological features; scalable; compatible with parallel drug testing | Epithelial-dominant; lack endogenous immune and stromal components; unable to capture tumor-immune interactions; limited suitability for modeling immunotherapy responses | Functional testing of chemotherapy and targeted agents; tumor cell-intrinsic response profiling | [33,96,108] |
| PDOTS | Preserve endogenous immune and stromal cells; enable short-term functional immune assays | Limited culture duration; lower scalability; protocol variability | Ex vivo assessment of immune-mediated tumor killing; immunotherapy response validation | [26,29,58,95,98] |
| PDTFs | Maintain intact tissue architecture and spatial immune organization; capture early immune reactivation | Very short viability window; low throughput; technically demanding | Early functional prediction of immunotherapy response; immune ecosystem interrogation | [28,62,64,65] |
PDOs AS PLATFORMS FOR TUMOR CELL-INTRINSIC FUNCTIONAL PRECISION ONCOLOGY IN HCC
Conceptual emergence of PDOs in liver cancer research
The emergence of PDOs in liver cancer research reflects a broader shift in preclinical oncology toward models that balance biological fidelity with experimental feasibility. Traditional in vitro systems are scalable but often fail to capture patient-specific tumor behavior, whereas in vivo platforms such as PDX models, despite greater physiological relevance, are limited by long establishment times and reduced clinical timeliness. PDOs therefore emerged as an intermediate platform capable of preserving essential tumor-intrinsic features while enabling rapid, parallelized functional interrogation.
The liver is particularly amenable to organoid modeling because hepatic epithelial cells display substantial plasticity and can undergo long-term expansion under defined culture conditions. Early studies showed that both normal and malignant liver tissues could be propagated as three-dimensional organoid cultures, providing the conceptual basis for applying this technology to HCC and related malignancies[68]. Importantly, PDOs were adopted in liver cancer research not simply because the technology was novel, but because they addressed a clear translational question: whether tumor cell-intrinsic drug sensitivities could be measured ex vivo within a clinically actionable timeframe. That question continues to shape the development and use of HCC-derived organoid systems[69].
Establishment strategies and biological fidelity of HCC-derived organoids
Tissue sources, culture conditions, and success rates
HCC-derived organoids have been established successfully from a range of tissue sources, including surgical specimens, biopsies, and metastatic lesions. Reported success rates generally range from about 30% to 70%, depending on tissue quality, tumor differentiation status, and culture conditions[70,71]. Tumors with higher proliferative capacity and stemness-associated transcriptional programs tend to form organoids more readily, suggesting an inherent biological bias in current PDO systems. Most protocols rely on extracellular matrix support and defined growth-factor supplementation to promote epithelial expansion while suppressing non-epithelial lineages. Although these conditions enable long-term propagation, they may also favor tumor-cell populations that adapt well to in vitro growth. This process can introduce clonal selection, enriching for highly proliferative or stem-like subclones while underrepresenting slower-growing populations from the original tumor[72]. Such shifts in cellular composition should be considered when interpreting functional drug-sensitivity assays and translational findings.
Genomic and histopathological concordance with parental tumors
A central requirement for the translational relevance of PDOs is their ability to recapitulate the molecular and histological features of their parental tumors. Multiple studies have shown that HCC-derived organoids retain key driver alterations, including mutations in TP53, CTNNB1 (catenin beta 1, encoding β-catenin), and TERT (telomerase reverse transcriptase), as well as copy-number changes and transcriptomic patterns that resemble those of the original tumors. This preservation should nevertheless be regarded as relative rather than complete, because long-term culture may favor expansion of specific subclones and may not fully maintain all complex structural or subclonal alterations present in the original lesion[73]. Histopathological analyses further support this fidelity, with PDOs reproducing tumor-specific architectural patterns and lineage-marker expression observed in matched patient tissues. Together, these findings establish PDOs as a reliable platform for studying tumor cell-intrinsic HCC biology at both genetic and phenotypic levels[74].
Functional drug sensitivity testing using HCC organoids
High-throughput drug screening and interpatient heterogeneity
One of the most compelling applications of PDOs is functional drug-sensitivity testing. Organoid cultures permit parallel evaluation of multiple agents across patient-derived samples and consistently reveal substantial interpatient heterogeneity in drug response[75]. This heterogeneity has been observed for both cytotoxic chemotherapy and targeted therapies commonly used in liver cancer[27]. In biliary tract cancer and liver cancer cohorts, PDO-based screening has uncovered response patterns that are not readily predicted by genomic data alone, underscoring the added value of functional assays[76,77].
Concordance between organoid drug response and clinical outcomes
The translational relevance of PDO-based drug testing is further supported by studies showing concordance between ex vivo organoid responses and patient clinical outcomes. In a representative biliary tract cancer study, drug sensitivities observed in PDOs matched patient responses in more than 90% of evaluable cases, with additional support from PDO-derived xenograft models[78]. Comparable large-scale validation studies in HCC remain limited, but emerging reports suggest similar trends, particularly for chemotherapy and targeted agents[54]. Discordant cases have also been described, reflecting both biological complexity and technical limitations. These discrepancies indicate that PDOs should not be viewed as universal predictors of therapy response, but rather as probabilistic functional readouts that must be interpreted within an appropriate biological context[79].
Organoid-based multi-omics profiling and drug response signatures
Beyond direct drug testing, PDOs are increasingly being combined with transcriptomic and proteogenomic profiling to dissect the molecular programs that underlie therapeutic response and resistance. Unlike bulk tumor specimens, which capture static molecular states at a single clinical time point, PDOs enable longitudinal, perturbation-aware profiling under controlled treatment conditions. This makes it possible to compare baseline, drug-exposed, and resistant states within matched genetic backgrounds and to obtain a dynamic view of tumor cell-intrinsic adaptation to therapy[80]. Transcriptomic studies of liver cancer PDOs have consistently identified treatment-associated reprogramming of signaling and metabolic pathways, including changes in cell-cycle regulation, stress responses, lipid and amino-acid metabolism, and lineage-associated transcriptional programs[81,82]. Many of these changes parallel patterns observed in matched patient tumors after therapy, supporting the biological relevance of PDO-derived molecular readouts. PDOs also permit analysis of adaptive transcriptional responses that are difficult to resolve in clinical samples because of limited tissue availability and confounding stromal and immune signals.
Proteogenomic and phosphoproteomic analyses complement transcriptomic profiling by providing direct information on pathway activation and functional effector states that cannot be inferred reliably from RNA expression alone[83,84]. Integration of protein abundance and phosphorylation dynamics in PDOs has revealed drug-induced rewiring of kinase networks, feedback activation of compensatory pathways, and post-transcriptional regulatory mechanisms that contribute to therapeutic escape. These approaches are especially informative for targeted therapies, for which pathway activity rather than gene expression per se often determines drug efficacy.
Together, these multi-omics studies highlight the value of PDOs as a mechanistically tractable system for linking functional drug-response phenotypes to underlying molecular states[85]. By enabling controlled and repeatable perturbation experiments coupled with deep molecular profiling, PDO-based transcriptomic and proteogenomic analyses help bridge the gap between descriptive clinical correlative studies and causal mechanistic investigation.
Building on these datasets, several groups have used organoid-derived drug-response profiles to generate gene-expression signatures or predictive panels for patient stratification. Some studies have reported encouraging performance in retrospective analyses, although accuracy varies substantially across tumor types, treatment settings, and clinical endpoints. For example, organoid-informed prediction has shown promise in selected malignancies, particularly colorectal cancer, whereas comparable large-scale validation in HCC remains limited[86]. It is important to note, however, that most organoid-derived signatures remain fundamentally correlative. Although functional assays provide a strong foundation for identifying response-associated molecular features, the predictive value of such panels ultimately depends on independent validation and prospective clinical testing.
Structural and functional boundaries of conventional HCC organoids
Despite their strengths, conventional HCC organoids remain intrinsically epithelial-dominant systems[87]. They lack critical components of the tumor microenvironment, including immune cells, fibroblasts, and vascular elements, all of which are increasingly recognized as key determinants of therapeutic response[37]. Consequently, PDOs are structurally unable to model immune-mediated mechanisms of action, particularly those relevant to immune checkpoint blockade[76].
This epithelial bias imposes a fundamental limitation on the use of PDOs in immunotherapy research. Although PDOs can capture tumor cell-intrinsic sensitivity to targeted therapy and chemotherapy, they cannot reliably predict response to ICIs or support rational testing of immunotherapy-based combinations. This limitation has driven the development of organotypic platforms that preserve endogenous immune components, including PDOTS and tumor-fragment systems.
In summary, PDOs are powerful platforms for modeling tumor cell-intrinsic biology and functional drug sensitivity in HCC, including aspects of tumor heterogeneity and cellular plasticity. HCC exhibits pronounced intratumoral heterogeneity and dynamic cellular plasticity during progression and under therapeutic pressure. Subpopulations with stem-like properties[88], partial epithelial-mesenchymal transition states, and drug-tolerant phenotypes may all contribute to adaptive resistance and tumor evolution[27,35]. These features also shape genomic and transcriptomic heterogeneity and may influence organoid establishment, because the presence and functional state of cancer stem-like cells likely affect PDO generation and long-term propagation. In this regard, organoid systems provide a valuable platform for studying tumor-cell plasticity, cancer stem-cell dynamics, and therapy-driven evolutionary processes, thereby offering additional insight into functional precision oncology.
At the same time, the absence of microenvironmental components defines a clear boundary to the applicability of conventional PDOs. They should therefore be viewed not as comprehensive models of therapeutic response, but as foundational elements within a broader ecosystem of patient-derived functional platforms. This perspective naturally leads to the exploration of organotypic systems that retain immune and stromal elements, as discussed in the following sections. Even these more advanced models still face important technical and biological challenges, including variable establishment success and incomplete representation of full tumor-microenvironment complexity, which must be addressed before routine clinical implementation.
ORGANOTYPIC TUMOR MODELS FOR FUNCTIONAL ASSESSMENT OF IMMUNOTHERAPY RESPONSE IN HCC
Rationale for organotypic modeling beyond conventional organoids
As discussed above, conventional PDOs are epithelial-dominant systems that lack key components of the tumor microenvironment. This limitation is especially consequential in HCC, where immune contexture and tumor-immune interactions critically influence responses to immune checkpoint blockade[76].
Clinical experience with PD-1/PD-L1 inhibitors underscores the complexity of immunotherapy response in HCC. Only a subset of patients achieves durable benefit, and these responses cannot be predicted reliably by tumor cell-intrinsic biomarkers alone. Instead, factors such as pre-existing tumor-infiltrating lymphocytes, immune exclusion, and spatially organized immune niches have emerged as major determinants of therapeutic outcome[89,90].
Organotypic tumor models were developed specifically to address this gap. By minimizing tissue dissociation and preserving endogenous cellular composition, these platforms enable ex vivo functional interrogation of immune responses within an intact tumor ecosystem, a capability that classical PDO systems do not provide.
PDOTS
Structural and biological distinction from classical PDOs
PDOTS differ fundamentally from conventional PDOs in both construction and biological scope. Rather than isolating epithelial tumor cells for long-term expansion, PDOTS are generated through limited enzymatic and mechanical dissociation of fresh tumor tissue, followed by enrichment of multicellular spheroids rather than complete single-cell dissociation. Whereas conventional PDO protocols typically rely on more extensive digestion to obtain epithelial-enriched populations for prolonged culture, PDOTS preparation is designed to preserve tumor cells together with resident immune populations, stromal elements, and extracellular matrix. In seminal microfluidic PDOTS studies, spheroids in the 40-100 μm range were further selected because this fraction retained local immune populations and was well suited to short-term ex vivo culture[57].
This distinction has important functional consequences. PDOTS retain endogenous CD8⁺ and CD4⁺ T cells, myeloid subsets, and tumor-associated fibroblasts during short-term culture, thereby preserving cellular interactions that are central to immune-mediated tumor control. Unlike PDOs, which primarily reflect tumor cell-intrinsic behavior, PDOTS provide a reduced yet biologically coherent representation of the tumor microenvironment.
Functional readouts and short-term immune responsiveness
A defining feature of PDOTS is their suitability for short-term functional assays. Because immune-cell viability and spatial organization are preserved only transiently, PDOTS are generally cultured for days rather than weeks. Within this limited window, however, they permit direct measurement of immune activation, cytokine production, and tumor-cell killing in response to therapeutic perturbation.
Several studies have shown that PDOTS can be used to assess ex vivo responses to immune checkpoint blockade. For example, exposure to PD-1 inhibitors can induce rapid changes in T-cell activation markers and effector-cytokine secretion, generating a functional readout of immune competence that is not captured by static molecular profiling[91].
Importantly, PDOTS are often incorporated as functional validation modules within broader translational studies, even when they are not presented as the primary experimental platform. In these settings, PDOTS-based assays have been used to corroborate immune responsiveness observed in vivo or inferred from transcriptomic analyses, underscoring their practical value as an intermediate functional system.
PDTFs: preserving native immune architecture
Concept and methodological features
PDTFs represent an extreme form of organotypic modeling characterized by minimal manipulation of tumor tissue. Small fragments of freshly resected tumors are maintained ex vivo with preservation of three-dimensional architecture, vascular remnants, and spatial immune organization[66].
Unlike PDOTS, which involve partial tissue reaggregation, PDTFs retain intact immune niches, including tertiary lymphoid structures and tumor-immune interfaces[92]. This architectural preservation allows interrogation of immune responses in their native spatial context, albeit over a very limited culture period.
Early immune reactivation as a predictor of clinical response
A landmark study using PDTFs showed that ex vivo PD-1 blockade could induce measurable immune reactivation within 24-48 h across multiple cancer types, including liver cancer[93]. Importantly, the magnitude of this early immune response, reflected by cytokine secretion and T-cell activation, correlated strongly with patient clinical outcomes[64].
This work provided direct functional evidence that responsiveness to immune checkpoint blockade is encoded, at least in part, within the pre-existing tumor immune ecosystem. It also identified distinct categories of nonresponsive tumors, including lesions with immune infiltration but without functionally tumor-reactive T cells, an observation with important implications for patient stratification.
The association between tertiary lymphoid structures and immune-reactivation capacity further underscored the importance of spatial immune organization in determining therapeutic response and reinforced the conceptual value of PDTFs as functional immunotherapy models.
Comparative positioning of PDOTS and PDTFs
Although PDOTS and PDTFs share the goal of preserving tumor-immune interactions, they occupy distinct positions within the organotypic modeling spectrum. PDOTS provide greater experimental flexibility, scalability, and compatibility with quantitative assays, making them suitable for functional screening and mechanistic studies[26,57]. PDTFs, in contrast, maximize biological fidelity at the expense of throughput and culture duration and are especially useful for probing early immune dynamics and spatially organized responses[92].
Different patient-derived tumor models may at times yield discordant predictions of therapeutic response. For instance, epithelial-dominant PDO systems may suggest sensitivity to targeted agents on the basis of tumor cell-intrinsic programs[33], whereas organotypic platforms such as PDOTS or PDTFs may reveal reduced efficacy once immune-mediated mechanisms or stromal interactions are considered[58]. Such discordance should not necessarily be interpreted as a technical failure; rather, it reflects the different biological dimensions captured by each model. These differences must therefore be interpreted in light of the specific processes represented by each system. At the same time, PDOTS and PDTFs still face practical challenges, including variable tissue quality, short culture duration, and limited throughput, all of which may constrain their current clinical scalability.
PDOTS and PDTFs should therefore be viewed as complementary rather than competing platforms. Together, they enable functional assessment of immunotherapy response across different levels of structural preservation and experimental control, addressing a major unmet need in precision oncology for HCC.
Implications for functional precision oncology in HCC
The emergence of organotypic tumor models marks a conceptual shift in preclinical HCC research, from static biomarker identification toward dynamic, patient-specific functional testing. By directly measuring immune responsiveness ex vivo, PDOTS and PDTFs provide a biologically grounded and clinically relevant means of interrogating therapeutic potential.
These platforms do not replace existing models, nor do they eliminate the need for molecular biomarkers. Instead, they add a functional layer of evidence that complements genomic and transcriptomic analysis and supports more integrative approaches to precision therapy development[66,94].
TRANSLATIONAL INTEGRATION OF PATIENT-DERIVED FUNCTIONAL TUMOR MODELS IN HCC
From experimental platforms to translational tools
Although PDO-based and organotypic tumor models were initially developed as experimental systems, their role in HCC research has increasingly shifted toward translational application. In many influential studies, these models are not presented as the central narrative element but are embedded as functional validation layers that complement in vivo models, molecular profiling, or clinical observations[59]. This pattern reflects an important conceptual transition. Rather than acting as standalone predictors, patient-derived functional models are increasingly used as contextual interrogators that test whether hypothesized mechanisms or biomarkers translate into measurable therapeutic responses at the tissue level. This embedded mode of use has become particularly prominent in studies of drug resistance and immunotherapy response, where static molecular data alone are insufficient to establish causality[95].
Organoid-based drug sensitivity testing informing clinical response
The most mature translational application of PDO models lies in drug-sensitivity testing for cytotoxic and targeted therapies. In biliary tract cancer, a landmark study established a large PDO biobank and reported case-level concordance between ex vivo drug-sensitivity patterns and observed clinical responses in 12 of 13 evaluable patient-PDO pairs, with additional confirmation in PDO-derived xenografts[96]. Representative studies using PDO, PDOTS, and PDTF models across different therapeutic settings are summarized in Table 2. In that study, concordance was assessed by comparing organoid-based chemotherapy sensitivity profiles, derived from dose-response and related viability readouts, with the corresponding patients’ treatment outcomes. This work provided one of the clearest demonstrations that organoid-based functional testing can inform therapeutic stratification. In HCC, similar approaches have been applied in smaller cohorts. Organoid-based testing has been used to evaluate sensitivity to sorafenib, lenvatinib, and other targeted agents and has revealed substantial interpatient heterogeneity that is not fully explained by genomic alterations alone[27,45]. Both concordant and discordant cases have been reported, underscoring that PDO-derived drug response should be interpreted as a probabilistic rather than deterministic predictor[86]. These findings suggest that PDO-based drug testing is most informative for therapies whose mechanisms are primarily tumor cell intrinsic, whereas its predictive value is lower for treatments that depend heavily on microenvironmental or immune-mediated effects.
Representative studies using PDO, PDOTS and PDTF models across different therapeutic applications
| Model | Cancer type | Therapy context | Sample size | Study purpose | References |
| PDO | HCC | Targeted therapy/Chemotherapy | 27 | Drug response evaluation | [27] |
| PDO | HCC, ICC, CHC, GBC | High-throughput drug screening (301 drugs) | 64 | Drug sensitivity testing; Functional validation; Mechanistic investigation | [83] |
| PDO | BTC | Chemotherapy drug screening (gemcitabine, cisplatin, 5-FU, etc.) | 61 | Drug sensitivity testing; Functional validation; Mechanistic investigation | [96] |
| PDO | HCC, ICC, CHC | High-throughput drug screening (76 drugs) | 65 | Drug sensitivity testing; Functional validation; Mechanistic investigation | [80] |
| PDOTS | HCC | Anlotinib + anti-PD-1 | 24 | Drug sensitivity testing; Immunotherapy response evaluation; Mechanistic investigation | [29] |
| PDOTS | HCC | Anti-PD-1 | 49 | Immunotherapy response evaluation | [95] |
| PDOTS | HCC | Anti-PD-1 ± 1C8 | 3-5 | Immunotherapy response evaluation | [59] |
| PDOTS | ICC | Gemcitabine ± SSA | 12 | Drug response evaluation | [94] |
| PDOTS | NSCLC | Targeted therapy + PD-1 | 2 | Immunotherapy response evaluation | [109] |
| PDTF | Melanoma/NSCLC/RCC | Anti-PD-1 | 37 | Immunotherapy response evaluation | [64] |
| PDTF | Glioblastoma | Anti-CD25 ± anti-PD-1 | 9 | Immunotherapy response evaluation | [62] |
A recurring observation in organoid-based studies is the dissociation between genomic similarity and functional response. Tumors sharing common driver mutations can display markedly different sensitivities to the same agents when tested ex vivo[97]. This phenomenon highlights a major limitation of genotype-centered precision oncology and underscores the complementary value of functional stratification. By compressing complex cellular states into measurable phenotypic outputs, such as viability, growth arrest, or cytotoxic response, organoid-based assays provide an integrative readout that captures regulatory and epigenetic influences not readily inferred from sequencing data. In this sense, functional organoid assays do not replace molecular profiling but instead contextualize it within a response-oriented framework.
Embedded use of PDOTS in immunotherapy-oriented studies
PDOTS as a functional validation layer rather than a central model
In contrast to PDOs, PDOTS are only rarely presented as the primary experimental system in translational immunotherapy studies. Their most influential use has instead been as embedded functional modules that validate immune responsiveness inferred from other data modalities. Several studies investigating immune regulation or therapeutic resistance have used PDOTS to assess immune-mediated tumor killing or T-cell activation ex vivo[29,95,98]. In these settings, PDOTS serve not as the central platform but as complementary assays that substantiate molecular or cellular findings. Their value lies in providing direct functional evidence that links mechanistic observations to immune behavior in an intact tumor-tissue context. This use pattern reflects a broader trend in translational oncology: organotypic models are valued less for scalability or long-term expansion than for their ability to answer narrowly defined functional questions that are otherwise difficult to address.
Linking ex vivo immune reactivity to in vivo and clinical observations
A major strength of PDOTS-based assays is their ability to connect ex vivo immune reactivity with in vivo or clinical observations. In several settings, immune activation detected in PDOTS, such as increased cytokine secretion or enhanced tumor-cell killing, has paralleled responses observed in animal models or patient samples treated with the same agents[26]. PDOTS assays have also been used to functionally support transcriptomic signatures of immune activation or resistance, providing an intermediate validation step between correlative molecular data and clinical interpretation. This integrative role is particularly valuable in HCC, where immune heterogeneity and spatial organization complicate direct extrapolation from bulk sequencing data.
PDTFs in early response assessment
PDTFs offer a distinct translational opportunity by enabling rapid functional assessment of intratumoral immune responsiveness under near-native tissue architecture. In a seminal study, Voabil et al. established an ex vivo PDTF platform and showed that the capacity of immune cells to be reactivated by PD-1 blockade
Although PDTFs are not yet suited to routine clinical implementation, their translational implications are considerable. Functional stratification based on early immune reactivation could support patient-enrichment strategies in immunotherapy trials, identify immune-nonresponsive tumor subtypes, and inform rational combination development. Rather than serving as standalone diagnostic tools, PDTF-based assays are best regarded as discovery-oriented and hypothesis-generating platforms that complement established biomarkers such as PD-L1 expression and TMB.
Integration with multi-omics and computational frameworks
An emerging application of patient-derived functional models is their integration with multi-omics data and computational analysis to improve the interpretability and biological grounding of response prediction. Although transcriptomic, proteomic, and other high-dimensional datasets have enabled the discovery of response-associated signatures, such approaches remain limited by their correlative nature and susceptibility to cohort-specific bias. Without direct functional validation, molecular signatures may reflect coincident associations rather than causal determinants of therapeutic response[100]. In this setting, functional readouts generated from PDOs, PDOTS, or PDTFs provide a critical anchoring layer by linking molecular states to experimentally validated response phenotypes. By supplying quantitative measures of drug sensitivity or immune activation under controlled conditions, these models offer reference points that can calibrate, constrain, and refine computational inference. Rather than competing with multi-omics-based prediction, functional assays complement computational frameworks by defining biologically meaningful labels against which molecular features can be interpreted.
Importantly, incorporation of functional data supports a shift from purely associative modeling toward more mechanistically informed predictive frameworks. Functional readouts can serve as training data for supervised learning, benchmarks for evaluating model generalizability, or filters for prioritizing response-associated features that are reproducibly linked to phenotype across experimental settings. From a computational perspective, several machine-learning approaches may be relevant. Supervised models, including regularized regression, random forests, and deep neural networks, can be used to associate organoid-derived functional readouts with transcriptomic, genomic, or proteomic features[101,102]. In parallel, multimodal integration frameworks designed to jointly analyze heterogeneous omics layers may help capture the complex relationships between tumor-intrinsic programs and treatment response. Practical implementation will require careful attention to dataset size, cross-cohort validation, and technical variability across both functional assays and multi-omics platforms.
Through this role, patient-derived functional models reduce reliance on indirect inference and strengthen the robustness of response signatures derived from high-dimensional molecular data. Taken together, this integrative strategy positions functional tumor models as enabling components of computational precision oncology. By grounding multi-omics analyses in experimentally validated phenotypes, they improve both the interpretability and the biological relevance of predictive models.
Practical considerations and translational perspective
Despite their considerable translational promise, patient-derived functional tumor models still face several practical constraints that limit large-scale clinical deployment. Tissue availability, variable culture success rates, cost, and the lack of standardized protocols all pose challenges to routine implementation. In addition, heterogeneity in tissue processing and assay readouts complicates reproducibility across centers, emphasizing the need for continued methodological harmonization and prospective validation.
Additional practical issues relate to tumor sampling and treatment history. Because most patient-derived models are established from relatively small biopsy or surgical specimens, intratumoral heterogeneity raises the possibility of sampling bias[29], whereby the sampled region may not fully capture the functional diversity of the entire tumor. In HCC, spatial variation in immune infiltration, stromal composition, and clonal architecture may all influence ex vivo drug-sensitivity profiles. Moreover, prior therapies, including targeted agents, locoregional interventions, or immunotherapy, may alter tumor-cell states and immune composition before tissue collection, thereby affecting both model-establishment efficiency and interpretation of functional drug responses.
Within these constraints, patient-derived functional models should be viewed not as standalone diagnostic tools but as complementary research platforms that refine clinical interpretation. By enabling direct measurement of therapeutic response at the tissue level, they provide functional insight that is difficult to obtain through molecular profiling or in vivo modeling alone. Their main value lies in bridging mechanistic hypotheses and clinical observations by supplying experimentally grounded evidence that strengthens the interpretation of response-associated biomarkers and therapeutic strategies.
Viewed in this way, patient-derived functional tumor models occupy a distinct translational niche within precision oncology. Rather than replacing established biomarkers or predictive frameworks, they act as integrative intermediates that connect biological understanding with functional validation and thereby support the rational development and interpretation of personalized treatment strategies in HCC.
PERSPECTIVES AND CONCLUSIONS: FUNCTIONAL TUMOR MODELS AS ANCHORS OF PRECISION ONCOLOGY IN HCC
Recent advances in PDO-based and organotypic tumor models reflect a broader conceptual transition in HCC research, from reliance on static molecular descriptors toward incorporation of functional, patient-specific response assessment. Platforms such as PDOs, PDOTS, and PDTFs provide experimentally tractable systems for interrogating therapeutic response at the tissue level while preserving key biological features of patient tumors.
Rather than replacing molecular biomarkers, functional tumor models should be viewed as complementary tools that contextualize genomic and transcriptomic data. By linking experimentally measurable response phenotypes to molecular features, these systems provide biologically grounded reference frameworks for interpreting treatment-associated signatures and refining predictive strategies.
Taken together, patient-derived functional tumor models provide an important bridge between mechanistic understanding and clinical application in HCC. When integrated with molecular profiling and computational analysis, these platforms have the potential to improve therapeutic stratification and support the development of more rational precision oncology strategies.
DECLARATIONS
Acknowledgement
The Graphical Abstract was created using Nano-Banana and Adobe Illustrator.
Authors’ contributions
Conceptualization, literature curation, formal analysis, visualization, writing-original draft, writing-review and editing: Han L
Conceptualization, literature curation, formal analysis, methodology, visualization, writing-original draft, writing-review and editing: Song F
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version 5.4, released 2026-03-20) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This study was supported by the National Natural Science Foundation of China grants (81871927). This study was supported by grants from Jiangsu Provincial Research Hospital (YJXYY202204-YSB07), Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX23_3423), and the Nantong Hepatobiliary and Pancreatic Surgery Disease Research Center Construction Project (HS2015001).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025. CA Cancer J Clin. 2025;75:10-45.
2. Chan SL, Sun HC, Xu Y, et al. The Lancet Commission on addressing the global hepatocellular carcinoma burden: comprehensive strategies from prevention to treatment. Lancet. 2025;406:731-78.
3. Yang X, Yang C, Zhang S, et al. Precision treatment in advanced hepatocellular carcinoma. Cancer Cell. 2024;42:180-97.
4. Pinter M, Jain RK, Duda DG. The current landscape of immune checkpoint blockade in hepatocellular carcinoma: a review. JAMA Oncol. 2021;7:113-23.
5. Grant MJ, Herbst RS, Goldberg SB. Selecting the optimal immunotherapy regimen in driver-negative metastatic NSCLC. Nat Rev Clin Oncol. 2021;18:625-44.
6. El-Khoueiry AB, Trojan J, Meyer T, et al. Nivolumab in sorafenib-naive and sorafenib-experienced patients with advanced hepatocellular carcinoma: 5-year follow-up from CheckMate 040. Ann Oncol. 2024;35:381-91.
7. Finn RS, Qin S, Ikeda M, et al.; IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894-905.
8. Lee MS, Ryoo BY, Hsu CH, et al.; GO30140 investigators. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol. 2020;21:808-20.
9. Zhu AX, Abbas AR, de Galarreta MR, et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med. 2022;28:1599-611.
10. Vesely MD, Zhang T, Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol. 2022;40:45-74.
11. Zhou KQ, Zhong YC, Song MF, et al. Distinct immune microenvironment of venous tumor thrombus in hepatocellular carcinoma at single-cell resolution. Hepatology. 2025;82:566-81.
12. Rizzo A, Ricci AD, Brandi G. Durvalumab: an investigational anti-PD-L1 antibody for the treatment of biliary tract cancer. Expert Opin Investig Drugs. 2021;30:343-50.
13. Cresswell GD, Nichol D, Spiteri I, et al. Mapping the breast cancer metastatic cascade onto ctDNA using genetic and epigenetic clonal tracking. Nat Commun. 2020;11:1446.
14. Kwee S, Chen X. Immunotherapy biomarkers for HCC: contemporary challenges and emerging opportunities. Hepatoma Res. 2022;8:32.
16. Yang C, Zhang S, Cheng Z, et al. Multi-region sequencing with spatial information enables accurate heterogeneity estimation and risk stratification in liver cancer. Genome Med. 2022;14:142.
17. Guo W, Sun YF, Shen MN, et al. Circulating tumor cells with stem-like phenotypes for diagnosis, prognosis, and therapeutic response evaluation in hepatocellular carcinoma. Clin Cancer Res. 2018;24:2203-13.
18. Cadamuro M, Fabris L, Zhang X, Strazzabosco M. Tumor microenvironment and immunology of cholangiocarcinoma. Hepatoma Res. 2022;8:11.
19. Khairnar R, Islam MA, Fleishman J, Kumar S. Shedding light on non-alcoholic fatty liver disease: pathogenesis, molecular mechanisms, models, and emerging therapeutics. Life Sci. 2023;312:121185.
20. Llovet JM, Pinyol R, Kelley RK, et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat Cancer. 2022;3:386-401.
21. Han X, Cai C, Deng W, et al. Landscape of human organoids: Ideal model in clinics and research. Innovation. 2024;5:100620.
22. Thorel L, Perréard M, Florent R, et al. Patient-derived tumor organoids: a new avenue for preclinical research and precision medicine in oncology. Exp Mol Med. 2024;56:1531-51.
23. Magré L, Verstegen MMA, Buschow S, van der Laan LJW, Peppelenbosch M, Desai J. Emerging organoid-immune co-culture models for cancer research: from oncoimmunology to personalized immunotherapies. J Immunother Cancer. 2023;11:e006290.
24. Abdolahi S, Ghazvinian Z, Muhammadnejad S, Saleh M, Asadzadeh Aghdaei H, Baghaei K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J Transl Med. 2022;20:206.
25. Choi Y, Lee S, Kim K, Kim SH, Chung YJ, Lee C. Studying cancer immunotherapy using patient-derived xenografts (PDXs) in humanized mice. Exp Mol Med. 2018;50:1-9.
26. Jenkins RW, Aref AR, Lizotte PH, et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018;8:196-215.
27. Broutier L, Mastrogiovanni G, Verstegen MM, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23:1424-35.
28. Liu S, Liang Z, Zhu L, et al. FXR-mediated antigen-specific CD8+ T cell enhances antitumor immunity in intrahepatic cholangiocarcinoma. J Immunother Cancer. 2025;13:e012259.
29. Song F, Hu B, Liang XL, et al. Anlotinib potentiates anti-PD1 immunotherapy via transferrin receptor-dependent CD8+ T-cell infiltration in hepatocellular carcinoma. Clin Transl Med. 2024;14:e1738.
30. Hwang S, Jung IJ, Lee KJ, Kim YK, Tak E. Cellular expansion of hepatocellular carcinoma organoids using decellularized liver scaffolds. Clin Transplant Res. 2025;39:366-77.
31. Li Z, Fu C, Chen Y, et al. D-amino acid oxidase suppresses hepatocellular carcinoma via oxidizing D-amino acids. J Transl Med. 2025;23:1359.
32. Xie M, Lin Z, Ji X, et al. FGF19/FGFR4-mediated elevation of ETV4 facilitates hepatocellular carcinoma metastasis by upregulating PD-L1 and CCL2. J Hepatol. 2023;79:109-25.
33. Liu L, Yu L, Li Z, Li W, Huang W. Patient-derived organoid (PDO) platforms to facilitate clinical decision making. J Transl Med. 2021;19:40.
34. Khawar MB, Wang Y, Majeed A, Afzal A, Haneef K, Sun H. Mini-organs with big impact: organoids in liver cancer studies. Oncol Res. 2023;31:677-88.
35. Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2020;17:139-52.
36. Parejo-Alonso B, Barneda D, Trabulo SMD, et al. PPARδ orchestrates a prometastatic metabolic response to microenvironmental cues in pancreatic cancer. Cancer Res. 2025;85:3275-91.
37. Airola C, Pallozzi M, Cesari E, et al. Hepatocellular-carcinoma-derived organoids: innovation in cancer research. Cells. 2024;13:1726.
38. Grattarola M, Pons N, Cannet F, et al. Establishment of a mouse hepatocellular carcinoma tumoroid panel recapitulating inter- and intra- heterogeneity for disease modelling and combinatorial drug discovery. Cell Commun Signal. 2025;23:410.
39. Song F, Hu B, Cheng JW, et al. Anlotinib suppresses tumor progression via blocking the VEGFR2/PI3K/AKT cascade in intrahepatic cholangiocarcinoma. Cell Death Dis. 2020;11:573.
40. Hu B, Li H, Guo W, et al. Establishment of a hepatocellular carcinoma patient-derived xenograft platform and its application in biomarker identification. Int J Cancer. 2020;146:1606-17.
41. Koga Y, Ochiai A. Systematic review of patient-derived xenograft models for preclinical studies of anti-cancer drugs in solid tumors. Cells. 2019;8:418.
42. Zhao B, Zhou Y, Cheng N, et al. Targeted inhibition of PDGFRA with avapritinib, markedly enhances lenvatinib efficacy in hepatocellular carcinoma in vitro and in vivo: clinical implications. J Exp Clin Cancer Res. 2025;44:139.
43. Tan M, Ye W, Liu Y, et al. Niclosamide prodrug enhances oral bioavailability and efficacy against hepatocellular carcinoma by targeting vasorin-TGFβ signalling. Br J Pharmacol. 2025;182:5517-35.
44. Zanella ER, Grassi E, Trusolino L. Towards precision oncology with patient-derived xenografts. Nat Rev Clin Oncol. 2022;19:719-32.
45. Xian L, Zhao P, Chen X, et al. Heterogeneity, inherent and acquired drug resistance in patient-derived organoid models of primary liver cancer. Cell Oncol. 2022;45:1019-36.
46. Yoshida GJ. Applications of patient-derived tumor xenograft models and tumor organoids. J Hematol Oncol. 2020;13:4.
47. Zhao Y, Shuen TWH, Toh TB, et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut. 2018;67:1845-54.
48. Cho K, Ro SW, Seo SH, et al. Genetically engineered mouse models for liver cancer. Cancers. 2019;12:14.
49. Wang T, Liu L, Fang J, et al. Conditional activation of c-MYC in distinct catecholaminergic cells drives development of neuroblastoma or somatostatinoma. Cancer Res. 2025;85:424-41.
50. Spranger S, Gajewski TF. A new paradigm for tumor immune escape: β-catenin-driven immune exclusion. J Immunother Cancer. 2015;3:43.
51. Zhu M, Kim J, Deng Q, et al. Loss of p53 and mutational heterogeneity drives immune resistance in an autochthonous mouse lung cancer model with high tumor mutational burden. Cancer Cell. 2023;41:1731-48.e8.
53. Moeini A, Haber PK, Sia D. Cell of origin in biliary tract cancers and clinical implications. JHEP Rep. 2021;3:100226.
54. Chen P, Zhang X, Ding R, et al. Patient-derived organoids can guide personalized-therapies for patients with advanced breast cancer. Adv Sci. 2021;8:e2101176.
55. Yáñez-Bartolomé M, Macarulla T, Tian TV. The potential of patient-derived organoids in precision medicine of biliary tract cancer. Cell Rep Med. 2023;4:101294.
56. Zhao JS, Shi S, Qu HY, et al. Glutamine synthetase licenses APC/C-mediated mitotic progression to drive cell growth. Nat Metab. 2022;4:239-53.
57. Hong HK, Yun NH, Jeong YL, et al. Establishment of patient-derived organotypic tumor spheroid models for tumor microenvironment modeling. Cancer Med. 2021;10:5589-98.
58. Aref AR, Campisi M, Ivanova E, et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip. 2018;18:3129-43.
59. Cai J, Song L, Zhang F, et al. Targeting SRSF10 might inhibit M2 macrophage polarization and potentiate anti-PD-1 therapy in hepatocellular carcinoma. Cancer Commun. 2024;44:1231-60.
60. Ibáñez-Molero S, Veldman J, Simon Nieto J, et al. Tumour-reactive heterotypic CD8 T cell clusters from clinical samples. Nature. 2026;649:467-76.
61. Lee H, Jeong S, Shin EC. Significance of bystander T cell activation in microbial infection. Nat Immunol. 2022;23:13-22.
62. Galvez-Cancino F, Navarrete M, Beattie G, et al. Regulatory T cell depletion promotes myeloid cell activation and glioblastoma response to anti-PD1 and tumor-targeting antibodies. Immunity. 2025;58:1236-53.e8.
63. Kaptein P, Slingerland N, Metoikidou C, et al. CD8-targeted IL2 unleashes tumor-specific immunity in human cancer tissue by reviving the dysfunctional T-cell pool. Cancer Discov. 2024;14:1226-51.
64. Pelly VS, Moeini A, Roelofsen LM, et al. Anti-inflammatory drugs remodel the tumor immune environment to enhance immune checkpoint blockade efficacy. Cancer Discov. 2021;11:2602-19.
65. Segura-Collar B, Cómitre-Mariano B, López DA, et al. The TRIB2-DNMT1 pathway generates an immune-cold microenvironment in glioblastoma, and its inhibition promotes immunotherapy. Cancer Immunol Res. 2025;13:1022-36.
66. Eguren-Santamaría I, Rodríguez I, Herrero-Martin C, et al. Short-term cultured tumor fragments to study immunotherapy combinations based on CD137 (4-1BB) agonism. Oncoimmunology. 2024;13:2373519.
67. McDonald PC, Chafe SC, Brown WS, et al. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology. 2019;157:823-37.
68. Ma H, Zhang VX, Tsui YM, et al. Targeting sterol O-acyltransferase 1 rewires fatty acid metabolism and uncovers immune vulnerability in hepatocellular carcinoma. Hepatology. 2025;Epub ahead of print.
69. Li L, Zhang Y, Zhou Y, et al. Quaternary nanoparticles enable sustained release of bortezomib for hepatocellular carcinoma. Hepatology. 2022;76:1660-72.
70. Dong R, Fei Y, He Y, et al. Lactylation-driven HECTD2 limits the response of hepatocellular carcinoma to lenvatinib. Adv Sci. 2025;12:e2412559.
71. Navarro P, Grazioso TP, Barquín A, et al. Multicenter study correlating molecular characteristics and clinical outcomes of cancer cases with patient-derived organoids. J Exp Clin Cancer Res. 2025;44:182.
72. Tan XP, He Y, Yang J, et al. Blockade of NMT1 enzymatic activity inhibits N-myristoylation of VILIP3 protein and suppresses liver cancer progression. Signal Transduct Target Ther. 2023;8:14.
73. Lee SH, Hu W, Matulay JT, et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell. 2018;173:515-28.e17.
74. Fang X, Shu L, Chen T, et al. Organoids derived from patients provide a new opportunity for research and individualized treatment of malignant peritoneal mesothelioma. Mol Cancer. 2024;23:12.
75. Wang Q, Guo F, Jin Y, Ma Y. Applications of human organoids in the personalized treatment for digestive diseases. Signal Transduct Target Ther. 2022;7:336.
76. Saito Y. Establishment of an organoid bank of biliary tract and pancreatic cancers and its application for personalized therapy and future treatment. J Gastroenterol Hepatol. 2019;34:1906-10.
77. Zou Z, Lin Z, Wu C, et al. Micro-engineered organoid-on-a-chip based on mesenchymal stromal cells to predict immunotherapy responses of HCC patients. Adv Sci. 2023;10:e2302640.
78. Fang H, Xu H, Yu J, Cao H, Li L. Human hepatobiliary organoids: recent advances in drug toxicity verification and drug screening. Biomolecules. 2024;14:794.
79. Yang H, Cheng J, Zhuang H, et al. Pharmacogenomic profiling of intra-tumor heterogeneity using a large organoid biobank of liver cancer. Cancer Cell. 2024;42:535-51.e8.
80. Ji S, Feng L, Fu Z, et al. Pharmaco-proteogenomic characterization of liver cancer organoids for precision oncology. Sci Transl Med. 2023;15:eadg3358.
81. Su SH, Mitani Y, Li T, et al. Lactate suppresses growth of esophageal adenocarcinoma patient-derived organoids through alterations in tumor NADH/NAD+ redox state. Biomolecules. 2024;14:1195.
82. Kong LR, Gupta K, Wu AJ, et al. A glycolytic metabolite bypasses “two-hit” tumor suppression by BRCA2. Cell. 2024;187:2269-87.e16.
83. Zhu Y, Tang S, Yuan Q, et al. Integrated characterization of hepatobiliary tumor organoids provides a potential landscape of pharmacogenomic interactions. Cell Rep Med. 2024;5:101375.
84. Lee H, Kim B, Park J, et al. Cancer stem cells: landscape, challenges and emerging therapeutic innovations. Signal Transduct Target Ther. 2025;10:248.
85. Nikolova MT, He Z, Seimiya M, et al. Fate and state transitions during human blood vessel organoid development. Cell. 2025;188:3329-48.e31.
86. Ooft SN, Weeber F, Dijkstra KK, et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med. 2019;11:eaay2574.
87. van Tienderen GS, Groot Koerkamp B, IJzermans JNM, van der Laan LJW, Verstegen MMA. Recreating tumour complexity in a dish: organoid models to study liver cancer cells and their extracellular environment. Cancers. 2019;11:1706.
88. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123:1911-8.
89. Jensen CE, Loaiza-Bonilla A, Bonilla-Reyes PA. Immune checkpoint inhibitors for hepatocellular carcinoma. Hepat Oncol. 2016;3:201-11.
90. Chen K, Liu J, Liu S, et al. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell. 2017;170:492-506.e14.
91. Nanaki S, Barmpalexis P, Iatrou A, Christodoulou E, Kostoglou M, Bikiaris DN. Risperidone controlled release microspheres based on poly(lactic acid)-poly(propylene adipate) novel polymer blends appropriate for long acting injectable formulations. Pharmaceutics. 2018;10:130.
92. Roelofsen LM, Voabil P, de Bruijn M, et al. Protocol for ex vivo culture of patient-derived tumor fragments. STAR Protoc. 2023;4:102282.
93. Jerby-Arnon L, Shah P, Cuoco MS, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175:984-97.e24.
94. Song F, Wang CG, Wang TL, et al. Enhancement of gemcitabine sensitivity in intrahepatic cholangiocarcinoma through Saikosaponin-a mediated modulation of the p-AKT/BCL-6/ABCA1 axis. Phytomedicine. 2024;133:155944.
95. Wu SY, Zhu YY, Sun JL, et al. Targeting tumor-intrinsic BCL9 reverses immunotherapy resistance by eliciting macrophage-mediated phagocytosis and antigen presentation. Nat Commun. 2025;16:10039.
96. Ren X, Huang M, Weng W, et al. Personalized drug screening in patient-derived organoids of biliary tract cancer and its clinical application. Cell Rep Med. 2023;4:101277.
97. Betge J, Rindtorff N, Sauer J, et al. The drug-induced phenotypic landscape of colorectal cancer organoids. Nat Commun. 2022;13:3135.
98. Song F, Wang CG, Mao JZ, et al. PANoptosis-based molecular subtyping and HPAN-index predicts therapeutic response and survival in hepatocellular carcinoma. Front Immunol. 2023;14:1197152.
99. Voabil P, de Bruijn M, Roelofsen LM, et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat Med. 2021;27:1250-61.
100. Papaccio F, García-Mico B, Gimeno-Valiente F, et al. “Proteotranscriptomic analysis of advanced colorectal cancer patient derived organoids for drug sensitivity prediction”. J Exp Clin Cancer Res. 2023;42:8.
101. Anand GM, Megale HC, Murphy SH, et al. Controlling organoid symmetry breaking uncovers an excitable system underlying human axial elongation. Cell. 2023;186:497-512.e23.
102. Nguyen NTB, Gevers S, Kok RNU, et al. Lactate controls cancer stemness and plasticity through epigenetic regulation. Cell Metab. 2025;37:903-19.e10.
103. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603-7.
104. Kapałczyńska M, Kolenda T, Przybyła W, et al. 2D and 3D cell cultures - a comparison of different types of cancer cell cultures. Arch Med Sci. 2018;14:910-9.
105. Guerrero-López P, Martín-Pardillos A, Bonet-Aleta J, et al. 2D versus 3D tumor-on-chip models to study the impact of tumor organization on metabolic patterns in vitro. Sci Rep. 2025;15:19506.
106. DuPage M, Jacks T. Genetically engineered mouse models of cancer reveal new insights about the antitumor immune response. Curr Opin Immunol. 2013;25:192-9.
107. Ireson CR, Alavijeh MS, Palmer AM, Fowler ER, Jones HJ. The role of mouse tumour models in the discovery and development of anticancer drugs. Br J Cancer. 2019;121:101-8.
108. Hu Y, Sui X, Song F, et al. Lung cancer organoids analyzed on microwell arrays predict drug responses of patients within a week. Nat Commun. 2021;12:2581.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.